Xinjiang Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Urumqi, 830011, China.
University of Chinese Academy of Sciences, Beijing, 100049, China.
BMC Bioinformatics. 2022 Jun 16;23(1):234. doi: 10.1186/s12859-022-04766-z.
Protein-protein interaction (PPI) plays an important role in regulating cells and signals. Despite the ongoing efforts of the bioassay group, continued incomplete data limits our ability to understand the molecular roots of human disease. Therefore, it is urgent to develop a computational method to predict PPIs from the perspective of molecular system.
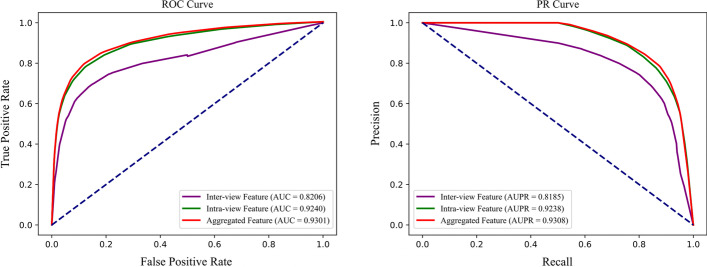
In this paper, a highly efficient computational model, MTV-PPI, is proposed for PPI prediction based on a heterogeneous molecular network by learning inter-view protein sequences and intra-view interactions between molecules simultaneously. On the one hand, the inter-view feature is extracted from the protein sequence by k-mer method. On the other hand, we use a popular embedding method LINE to encode the heterogeneous molecular network to obtain the intra-view feature. Thus, the protein representation used in MTV-PPI is constructed by the aggregation of its inter-view feature and intra-view feature. Finally, random forest is integrated to predict potential PPIs.
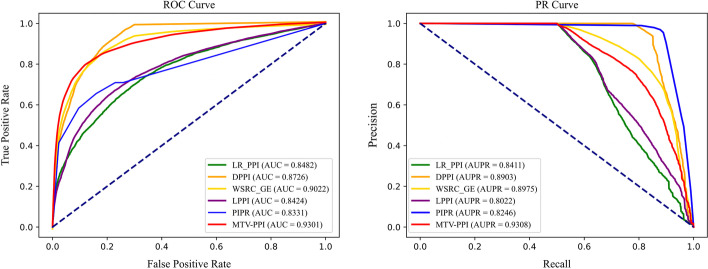
To prove the effectiveness of MTV-PPI, we conduct extensive experiments on a collected heterogeneous molecular network with the accuracy of 86.55%, sensitivity of 82.49%, precision of 89.79%, AUC of 0.9301 and AUPR of 0.9308. Further comparison experiments are performed with various protein representations and classifiers to indicate the effectiveness of MTV-PPI in predicting PPIs based on a complex network.
The achieved experimental results illustrate that MTV-PPI is a promising tool for PPI prediction, which may provide a new perspective for the future interactions prediction researches based on heterogeneous molecular network.
蛋白质-蛋白质相互作用(PPI)在调节细胞和信号方面起着重要作用。尽管生物测定组不断努力,但数据的持续不完整限制了我们理解人类疾病分子根源的能力。因此,迫切需要开发一种从分子系统角度预测 PPI 的计算方法。
本文提出了一种基于异构分子网络的高效计算模型 MTV-PPI,通过同时学习跨视图蛋白质序列和分子内视图相互作用来预测 PPI。一方面,通过 k-mer 方法从蛋白质序列中提取跨视图特征。另一方面,我们使用流行的嵌入方法 LINE 对异构分子网络进行编码,以获得内视图特征。因此,MTV-PPI 中使用的蛋白质表示是通过其跨视图特征和内视图特征的聚合构建的。最后,集成随机森林来预测潜在的 PPI。
为了证明 MTV-PPI 的有效性,我们在收集的异构分子网络上进行了广泛的实验,准确率为 86.55%,灵敏度为 82.49%,精度为 89.79%,AUC 为 0.9301,AUPR 为 0.9308。进一步进行了各种蛋白质表示和分类器的比较实验,以表明 MTV-PPI 在基于复杂网络预测 PPI 方面的有效性。
实验结果表明,MTV-PPI 是一种很有前途的 PPI 预测工具,它可能为未来基于异构分子网络的相互作用预测研究提供新的视角。