Su Xiao-Rui, You Zhu-Hong, Hu Lun, Huang Yu-An, Wang Yi, Yi Hai-Cheng
Xinjiang Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Ürümqi, China.
University of Chinese Academy of Sciences, Beijing, China.
Front Genet. 2021 Feb 26;12:635451. doi: 10.3389/fgene.2021.635451. eCollection 2021.
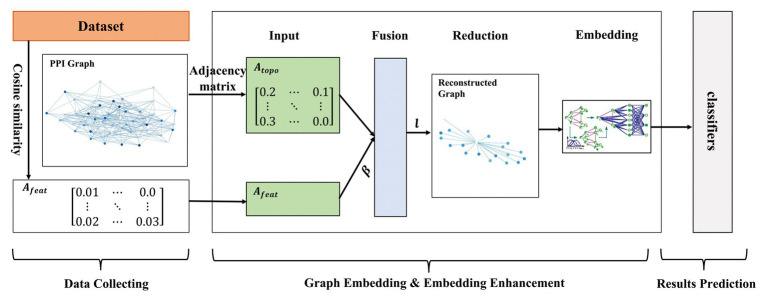
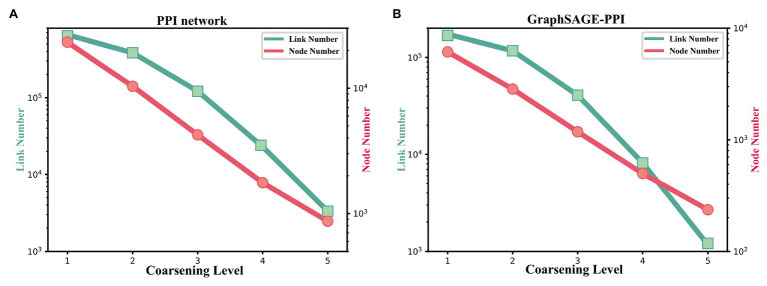
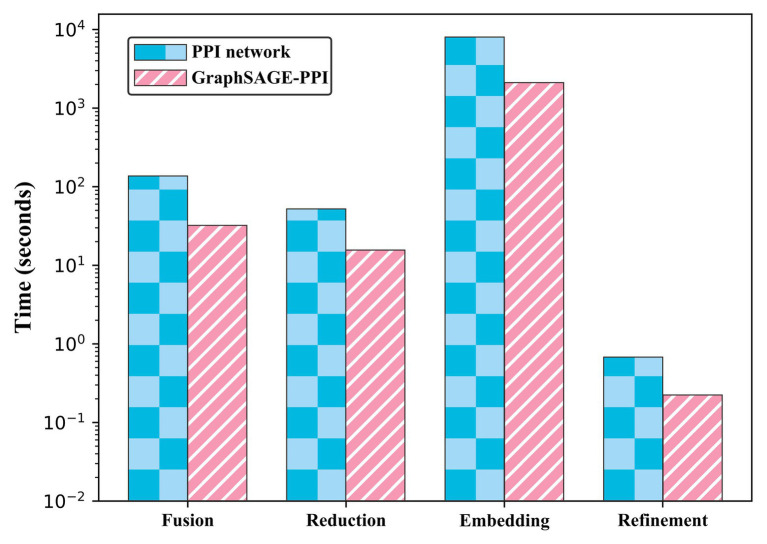
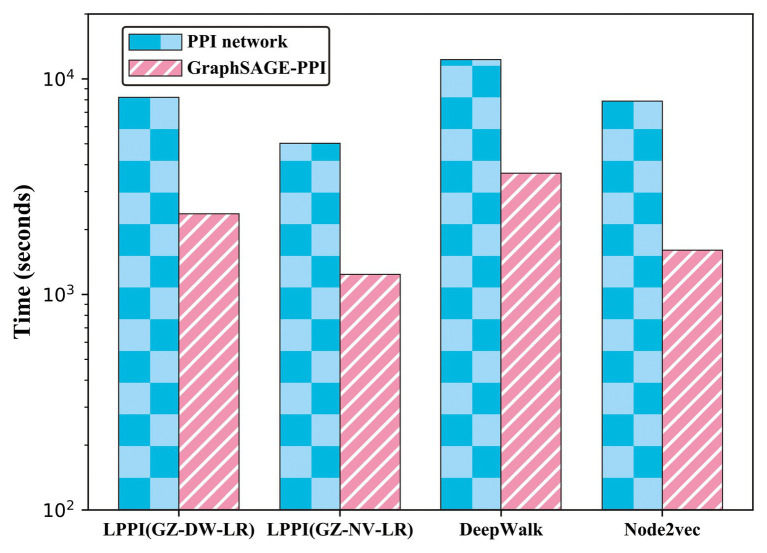
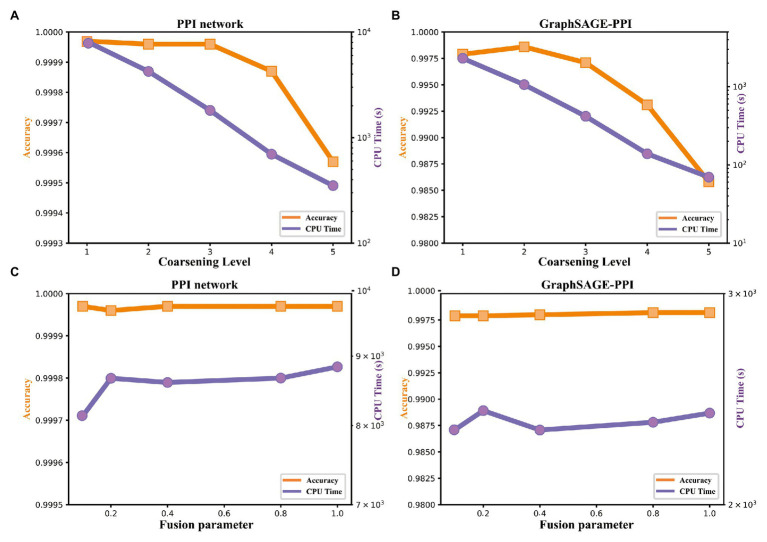
Protein-protein interaction (PPI) is the basis of the whole molecular mechanisms of living cells. Although traditional experiments are able to detect PPIs accurately, they often encounter high cost and require more time. As a result, computational methods have been used to predict PPIs to avoid these problems. Graph structure, as the important and pervasive data carriers, is considered as the most suitable structure to present biomedical entities and relationships. Although graph embedding is the most popular approach for graph representation learning, it usually suffers from high computational and space cost, especially in large-scale graphs. Therefore, developing a framework, which can accelerate graph embedding and improve the accuracy of embedding results, is important to large-scale PPIs prediction. In this paper, we propose a multi-level model LPPI to improve both the quality and speed of large-scale PPIs prediction. Firstly, protein basic information is collected as its attribute, including positional gene sets, motif gene sets, and immunological signatures. Secondly, we construct a weighted graph by using protein attributes to calculate node similarity. Then GraphZoom is used to accelerate the embedding process by reducing the size of the weighted graph. Next, graph embedding methods are used to learn graph topology features from the reconstructed graph. Finally, the linear Logistic Regression (LR) model is used to predict the probability of interactions of two proteins. LPPI achieved a high accuracy of 0.99997 and 0.9979 on the PPI network dataset and GraphSAGE-PPI dataset, respectively. Our further results show that the LPPI is promising for large-scale PPI prediction in both accuracy and efficiency, which is beneficial to other large-scale biomedical molecules interactions detection.
蛋白质-蛋白质相互作用(PPI)是活细胞整个分子机制的基础。尽管传统实验能够准确检测PPI,但它们往往成本高昂且耗时较长。因此,人们采用计算方法来预测PPI以避免这些问题。图结构作为重要且普遍存在的数据载体,被认为是呈现生物医学实体及其关系的最合适结构。虽然图嵌入是图表示学习中最流行的方法,但它通常存在高计算成本和高空间成本的问题,尤其是在大规模图中。因此,开发一个能够加速图嵌入并提高嵌入结果准确性的框架对于大规模PPI预测至关重要。在本文中,我们提出了一种多层次模型LPPI,以提高大规模PPI预测的质量和速度。首先,收集蛋白质的基本信息作为其属性,包括位置基因集、基序基因集和免疫特征。其次,我们利用蛋白质属性构建加权图以计算节点相似度。然后使用GraphZoom通过减小加权图的大小来加速嵌入过程。接下来,使用图嵌入方法从重建后的图中学习图拓扑特征。最后,使用线性逻辑回归(LR)模型预测两种蛋白质相互作用的概率。LPPI在PPI网络数据集和GraphSAGE-PPI数据集上分别达到了0.99997和0.9979的高精度。我们的进一步结果表明,LPPI在大规模PPI预测的准确性和效率方面都很有前景,这对其他大规模生物医学分子相互作用检测有益。