Araújo Leonardo Pereira de, Dias Maria Eduarda Carvalho, Scodeler Gislaine Cristina, Santos Ana de Souza, Soares Letícia Martins, Corsetti Patrícia Paiva, Padovan Ana Carolina Barbosa, Silveira Nelson José de Freitas, de Almeida Leonardo Augusto
Laboratory of Molecular Biology of Microorganisms, Federal University of Alfenas, Alfenas, Minas Gerais, Brazil.
Laboratory of Molecular Modeling and Computer Simulation, Federal University of Alfenas, Alfenas, Minas Gerais, Brazil.
Immunoinformatics (Amst). 2022 Sep;7:100015. doi: 10.1016/j.immuno.2022.100015. Epub 2022 Jun 11.
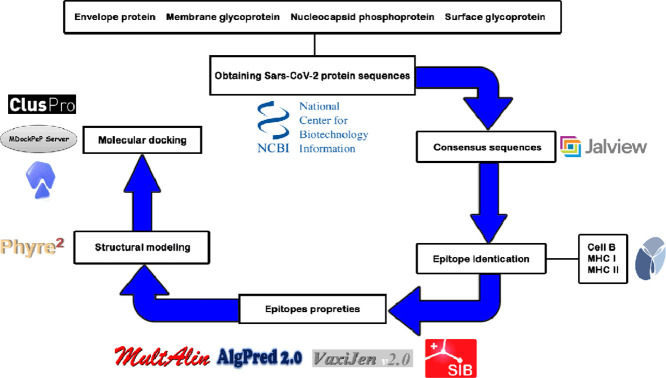
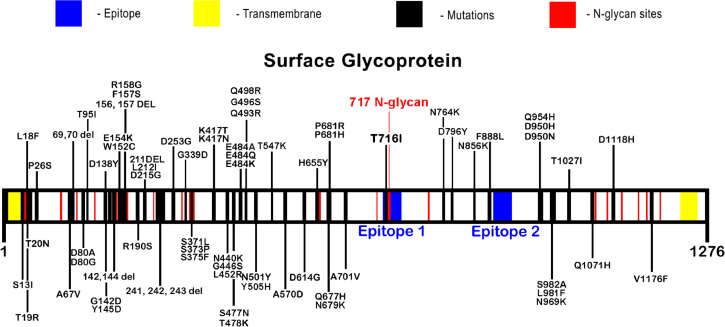
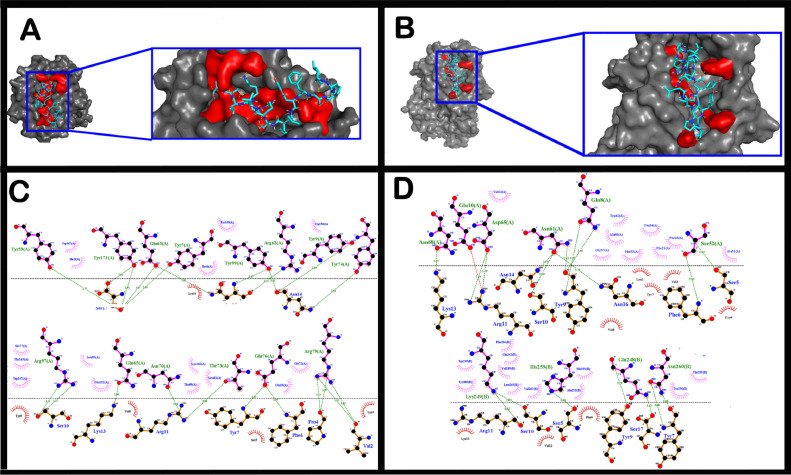
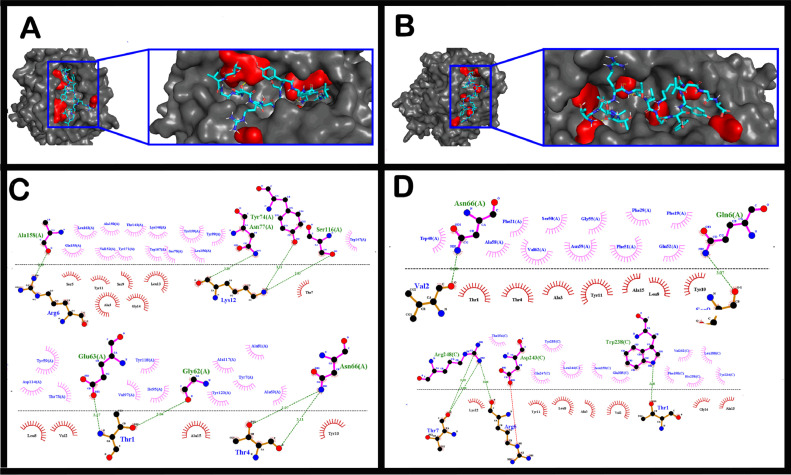
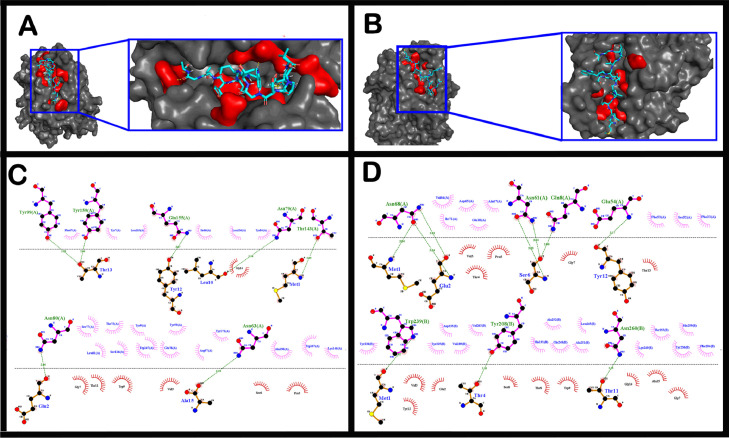
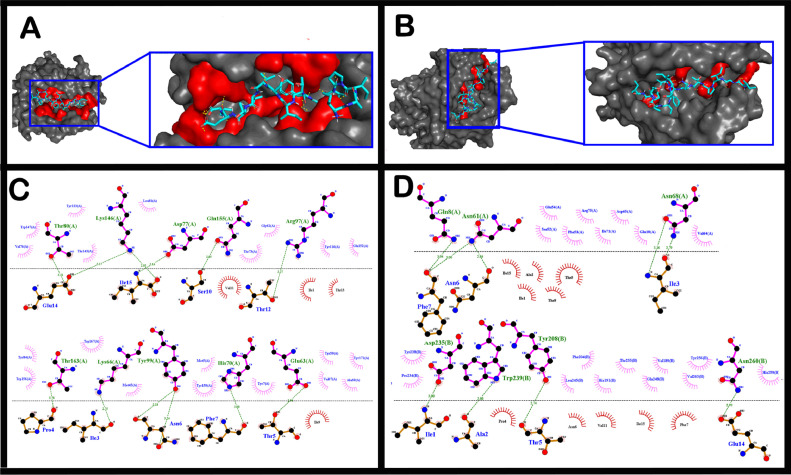
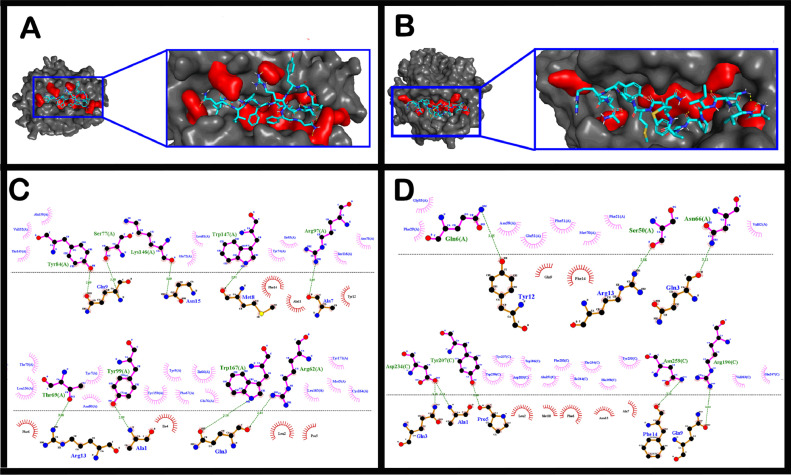
The short time between the first cases of COVID-19 and the declaration of a pandemic initiated the search for ways to stop the spread of SARS-CoV-2. There are great expectations regarding the development of effective vaccines that protect against all variants, and in the search for it, we hypothesized the obtention of a predicted rational immunogenic peptide from structural components of SARS-CoV-2 might help the vaccine research direction. In the search for a candidate of an immunogenic peptide of the SARS-CoV-2 envelope (E), membrane (M), nucleocapsid (N), or spike (S) proteins, we access the predicted sequences of each protein after the genome sequenced worldwide. We obtained the consensus amino acid sequences of about 14,441 sequences of each protein of each continent and the worldwide consensus sequence. For epitope identification and characterization from each consensus structural protein related to MHC-I or MHC-II interaction and B-cell receptor recognition, we used the IEDB reaching 68 epitopes to E, 174 to M, 245 to N, and 833 to S proteins. To select an epitope with the highest probability of binding to the MHC or BCR, all epitopes of each consensus sequence were aligned. The curation indicated 1, 4, 8, and 21 selected epitopes for E, M, N, and S proteins, respectively. Those epitopes were tested in silico for antigenicity obtaining 16 antigenic epitopes. Physicochemical properties and allergenicity evaluation of the obtained epitopes were done. Ranking the results, we obtained one epitope of each protein except for the S protein that presented two epitopes after the selection. To check the 3D position of each selected epitope in the protein structure, we used molecular homology modeling. Afterward, each selected epitope was evaluated by molecular docking to reference MHC-I or MHC-II allelic protein sequences. Taken together, the results obtained in this study showed a rational search for a putative immunogenic peptide of SARS-CoV-2 structural proteins that can improve vaccine development using in silico approaches. The epitopes selected represent the most conserved sequence of new coronavirus and may be used in a variety of vaccine development strategies since they are also presented in the described variants of SARS-CoV-2.
从首例新冠病毒病例出现到宣布大流行之间的时间很短,这促使人们寻找阻止严重急性呼吸综合征冠状病毒2(SARS-CoV-2)传播的方法。人们对开发能抵御所有变种的有效疫苗寄予厚望,在寻找此类疫苗的过程中,我们推测从SARS-CoV-2的结构成分中获取预测的合理免疫原性肽可能有助于疫苗研究方向。在寻找SARS-CoV-2包膜(E)、膜(M)、核衣壳(N)或刺突(S)蛋白的免疫原性肽候选物时,我们在全球基因组测序后获取了每种蛋白的预测序列。我们获得了每个大陆每种蛋白约14441个序列的共有氨基酸序列以及全球共有序列。为了从与主要组织相容性复合体I类(MHC-I)或MHC-II相互作用以及B细胞受体识别相关的每种共有结构蛋白中鉴定和表征表位,我们使用了免疫表位数据库(IEDB),分别得到了E蛋白的68个表位、M蛋白的174个表位、N蛋白的245个表位和S蛋白的833个表位。为了选择与MHC或B细胞受体结合概率最高的表位,对每个共有序列的所有表位进行了比对。筛选表明,E、M、N和S蛋白分别有1个、4个、8个和21个选定表位。对这些表位进行了计算机模拟抗原性测试,得到16个抗原性表位。对获得的表位进行了理化性质和致敏性评估。对结果进行排序后,除S蛋白在筛选后有两个表位外,每种蛋白都得到了一个表位。为了检查每个选定表位在蛋白质结构中的三维位置,我们使用了分子同源建模。之后,通过分子对接将每个选定表位与参考MHC-I或MHC-II等位蛋白序列进行评估。综上所述,本研究获得的结果表明,通过计算机模拟方法对SARS-CoV-2结构蛋白的假定免疫原性肽进行了合理搜索,这有助于改进疫苗开发。所选表位代表了新冠病毒最保守的序列,由于它们也存在于所描述的SARS-CoV-2变种中,因此可用于多种疫苗开发策略。