Department of Neuropediatrics, Astrid Lindgren Children´s Hospital, Karolinska University Hospital, Stockholm, Sweden.
Department of Women's and Children's Health, Karolinska Institute, Stockholm, Sweden.
Drug Des Devel Ther. 2022 Jun 16;16:1865-1883. doi: 10.2147/DDDT.S214174. eCollection 2022.
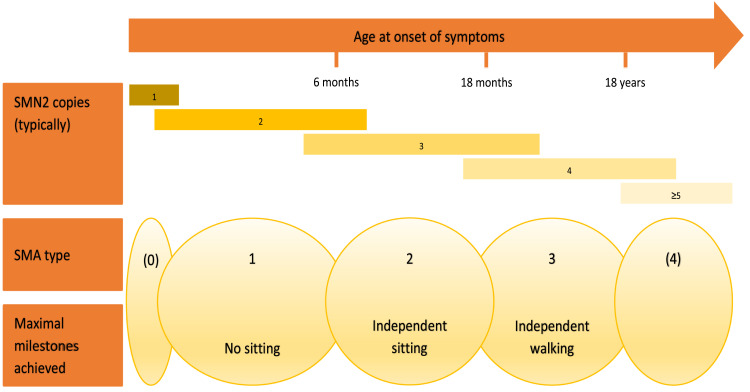
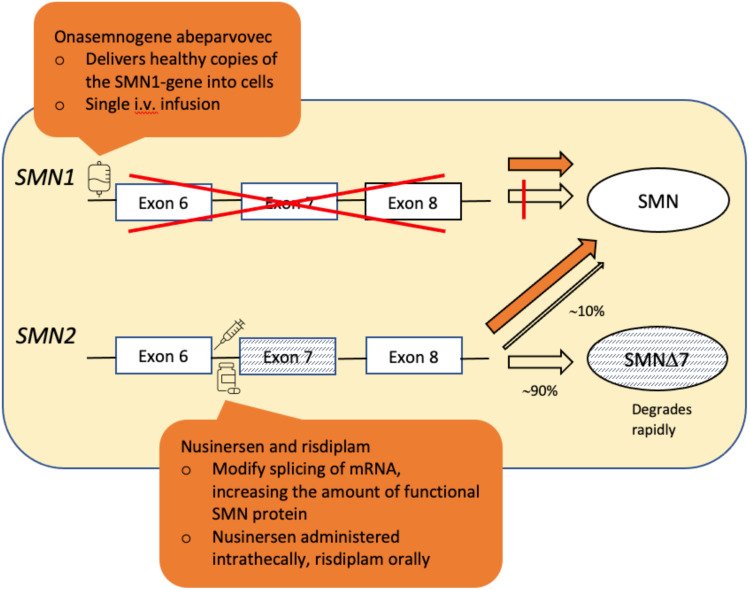
SMA (5q SMA) is an autosomal recessive neuromuscular disease with an estimated incidence of approximately 1 in 11,000 live births, characterized by progressive degeneration and loss of α-motor neurons in the spinal cord and brain stem, resulting in progressive muscle weakness. The disease spectrum is wide, from a serious congenital to a mild adult-onset disease. SMA is caused by biallelic mutations in the gene and disease severity is modified primarily by copy number. Before the advent of specific disease altering treatments, SMA was the second most common fatal autosomal recessive disorder after cystic fibrosis and the most common genetic cause of infant mortality. Nusinersen, risdiplam, and onasemnogene abeparvovec are presently the only approved disease modifying therapies for SMA, and the aim of this review is to discuss their mode of action, effects, safety concerns, and results from real-world experience. All exert their action by increasing the level of SMN protein in lower motor neuron. Nusinersen and risdiplam by modifying the SMN2 gene product, and onasemnogene abeparvovec by delivering SMN1 gene copies into cells. All have an established clinical efficacy. An important feature shared by all three is that early intervention is associated with a better treatment outcome, such that in cases where treatment is initiated in an early pre-symptomatic period, it may result in normal - or almost normal - motor development. Thus, early diagnosis followed by swift initiation of treatment is fundamental for the treatment response and consequently long-term prognosis in SMA type 1, and probably SMA type 2. The same principle similarly applies to the milder phenotypes. All three therapies are relatively novel, with risdiplam being the latest addition. Except for nusinersen, real-world data are still scarce, and long-term data are quite naturally lacking.
脊髓性肌萎缩症(5q SMA)是一种常染色体隐性神经肌肉疾病,估计发病率约为每 11000 例活产儿中有 1 例,其特征是脊髓和脑干中的α运动神经元进行性退化和丧失,导致进行性肌肉无力。疾病谱很广,从严重的先天性疾病到轻度成人发病疾病。SMA 是由 基因的双等位基因突变引起的,疾病的严重程度主要由 拷贝数修饰。在特定的疾病改变治疗方法出现之前,SMA 是继囊性纤维化之后第二常见的致命常染色体隐性遗传病,也是婴儿死亡的最常见遗传原因。诺西那生钠、利司扑兰和onasemnogene abeparvovec 是目前唯一批准用于治疗 SMA 的疾病修正疗法,本综述的目的是讨论它们的作用机制、效果、安全性问题以及真实世界经验的结果。所有这些都通过增加下运动神经元中 SMN 蛋白的水平来发挥作用。诺西那生钠和利司扑兰通过修饰 SMN2 基因产物,onasemnogene abeparvovec 通过将 SMN1 基因拷贝递送到细胞中。所有这些都有明确的临床疗效。这三种药物有一个重要的共同特征,即早期干预与更好的治疗效果相关,例如,在早期无症状期开始治疗的情况下,可能导致正常或几乎正常的运动发育。因此,早期诊断后迅速开始治疗对于 SMA 1 型的治疗反应和长期预后至关重要,可能对 SMA 2 型也同样如此。同样的原则也适用于更温和的表型。所有三种疗法都相对较新,risdiplam 是最新加入的。除了 nusinersen,真实世界的数据仍然很少,而且自然缺乏长期数据。