Department of Electrical Engineering and Computer Science, Informatics Institute, Christopher S. Bond Life Sciences Center, University of Missouri, Columbia, MO 65211, USA.
Fujian Provincial Key Laboratory of Innovative Drug Target Research, School of Pharmaceutical Sciences, Xiamen University, Xiamen 361000, China.
Biomolecules. 2022 May 25;12(6):746. doi: 10.3390/biom12060746.
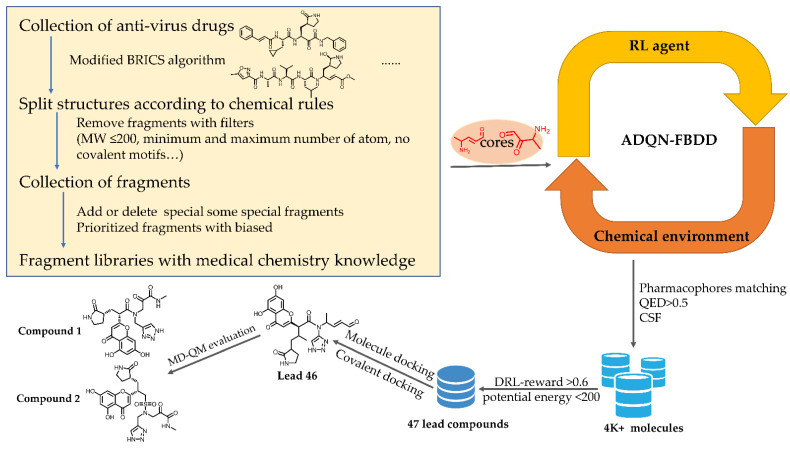
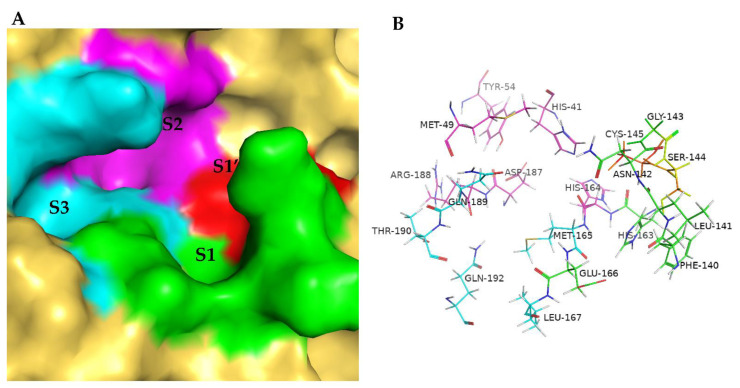
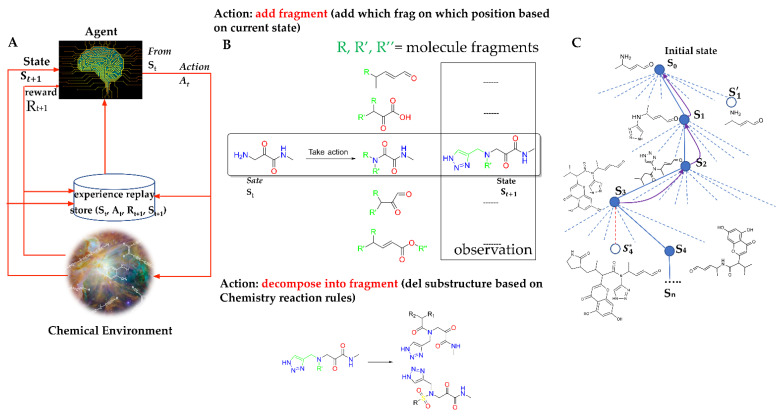
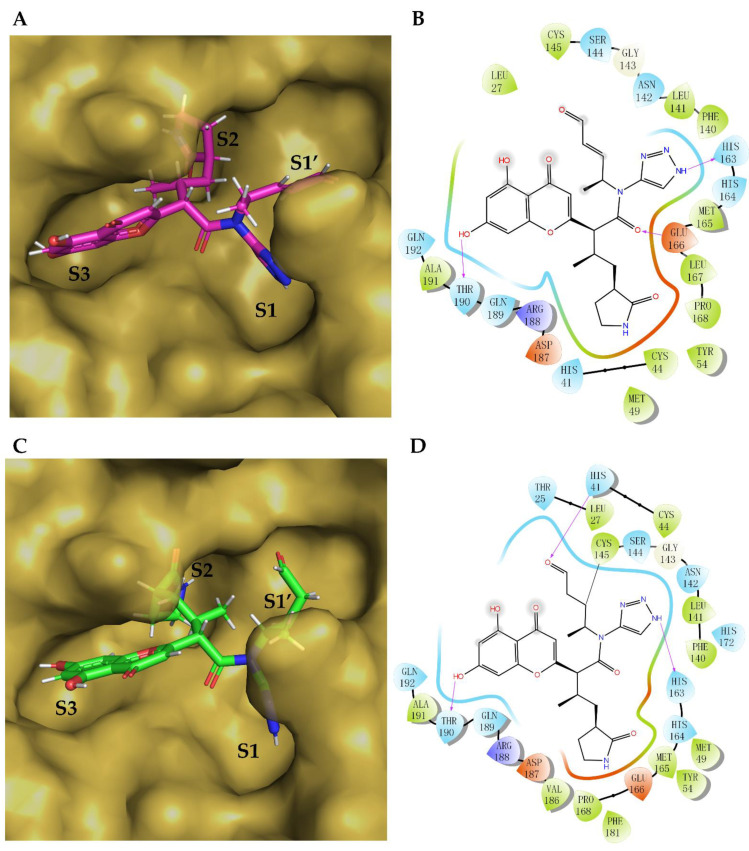
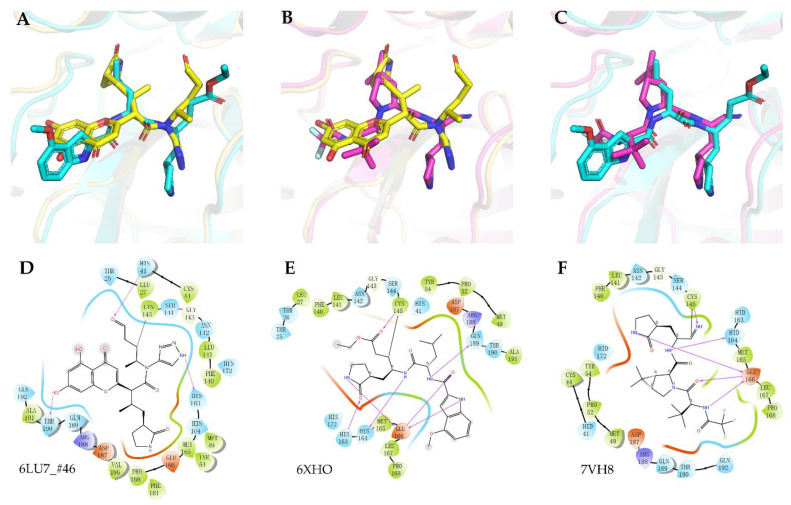
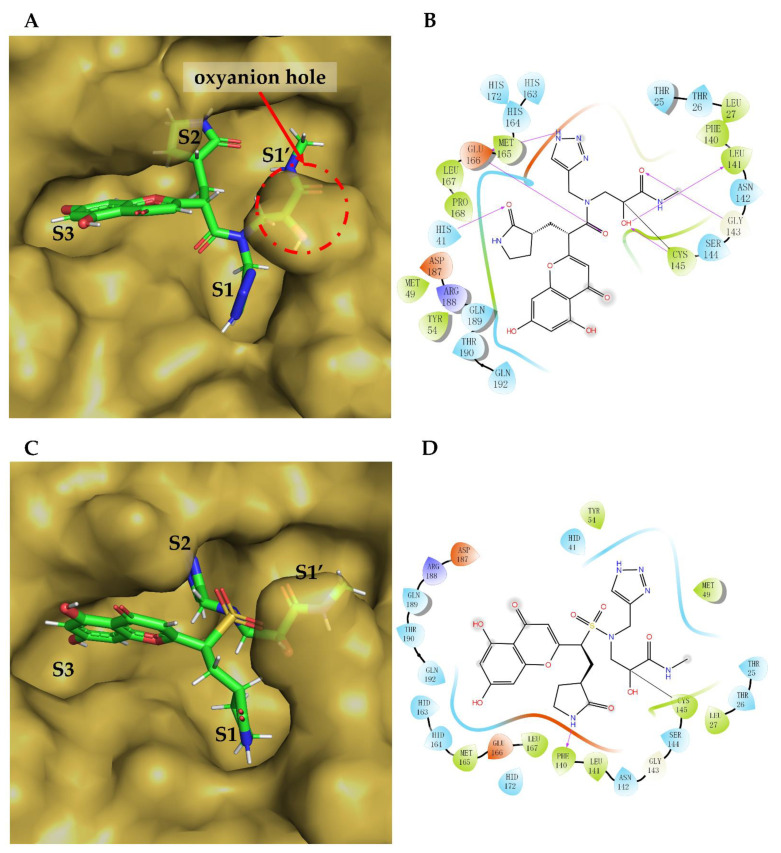
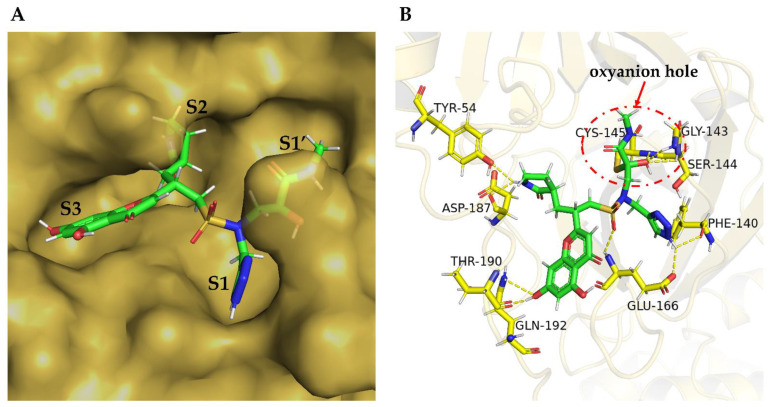
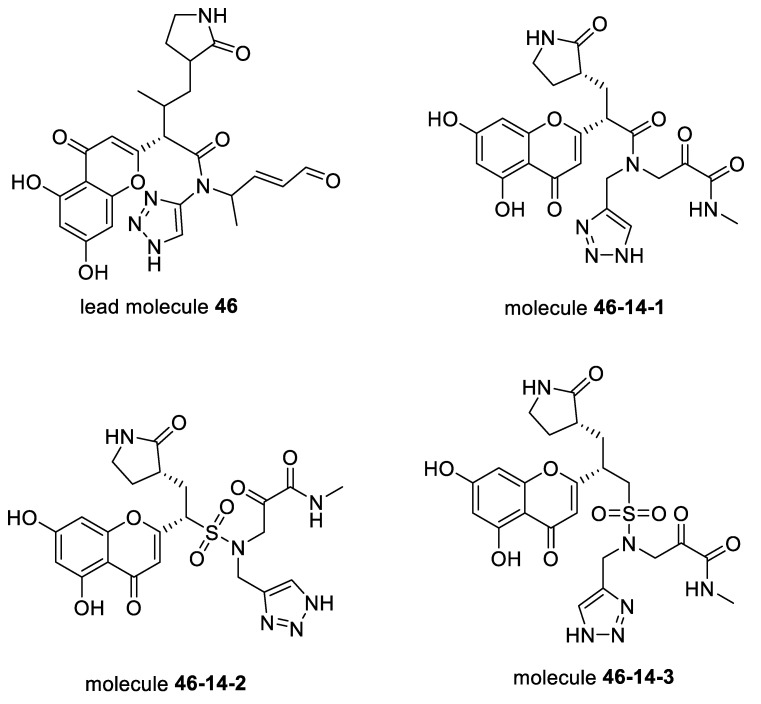
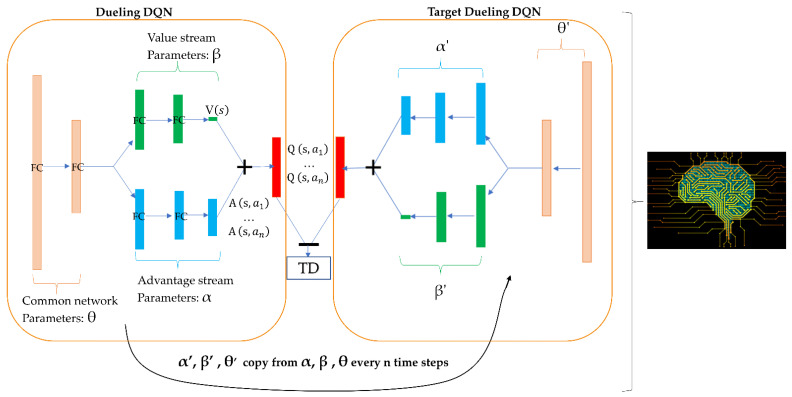
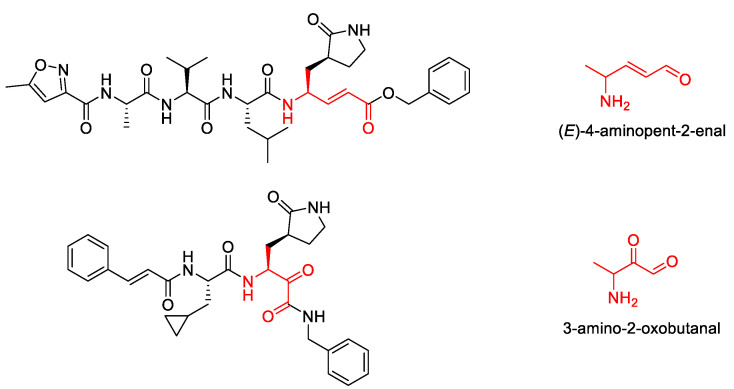
The drug repurposing of known approved drugs (e.g., lopinavir/ritonavir) has failed to treat SARS-CoV-2-infected patients. Therefore, it is important to generate new chemical entities against this virus. As a critical enzyme in the lifecycle of the coronavirus, the 3C-like main protease (3CLpro or Mpro) is the most attractive target for antiviral drug design. Based on a recently solved structure (PDB ID: 6LU7), we developed a novel advanced deep Q-learning network with a fragment-based drug design (ADQN-FBDD) for generating potential lead compounds targeting SARS-CoV-2 3CLpro. We obtained a series of derivatives from the lead compounds based on our structure-based optimization policy (SBOP). All of the 47 lead compounds obtained directly with our AI model and related derivatives based on the SBOP are accessible in our molecular library. These compounds can be used as potential candidates by researchers to develop drugs against SARS-CoV-2.
已知批准药物(例如洛匹那韦/利托那韦)的药物再利用未能治疗 SARS-CoV-2 感染患者。因此,生成针对这种病毒的新化学实体非常重要。作为冠状病毒生命周期中的关键酶,3C 样主蛋白酶(3CLpro 或 Mpro)是抗病毒药物设计最具吸引力的靶标。基于最近解决的结构(PDB ID:6LU7),我们开发了一种新的基于片段的药物设计(ADQN-FBDD)的新型高级深度 Q 学习网络,用于生成针对 SARS-CoV-2 3CLpro 的潜在先导化合物。我们根据基于结构的优化策略(SBOP)从先导化合物中获得了一系列衍生物。我们的 AI 模型直接获得的 47 种先导化合物以及基于 SBOP 的相关衍生物都可在我们的分子库中获得。这些化合物可以作为潜在的候选药物,由研究人员用于开发针对 SARS-CoV-2 的药物。