Institute of Cereal and Oil Crops, Hebei Academy of Agricultural and Forestry Sciences/Hebei Laboratory of Crop Genetics and Breeding, Shijiazhuang 050035, China.
Biobin Data Sciences, Changsha 410221, China.
Plant Commun. 2022 Nov 14;3(6):100352. doi: 10.1016/j.xplc.2022.100352. Epub 2022 Jun 26.
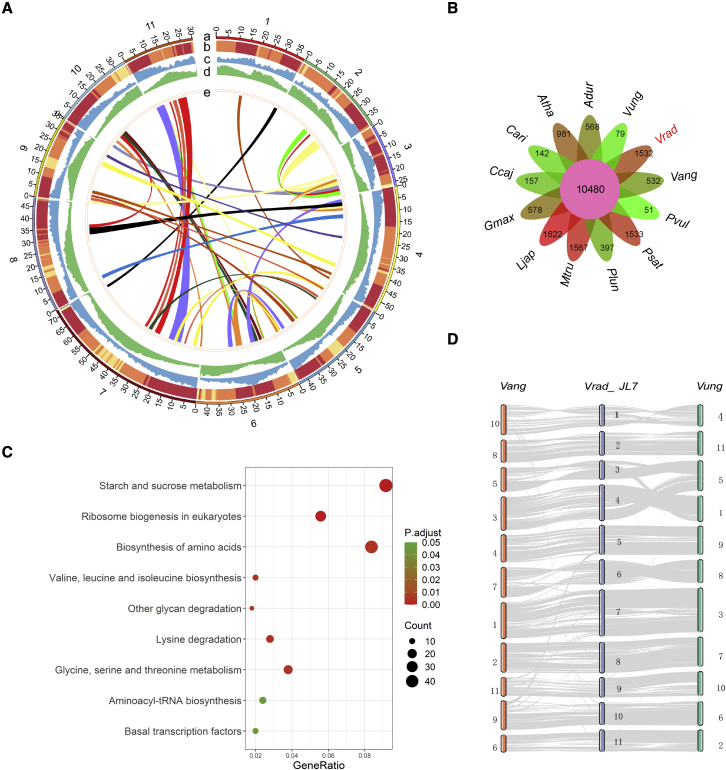
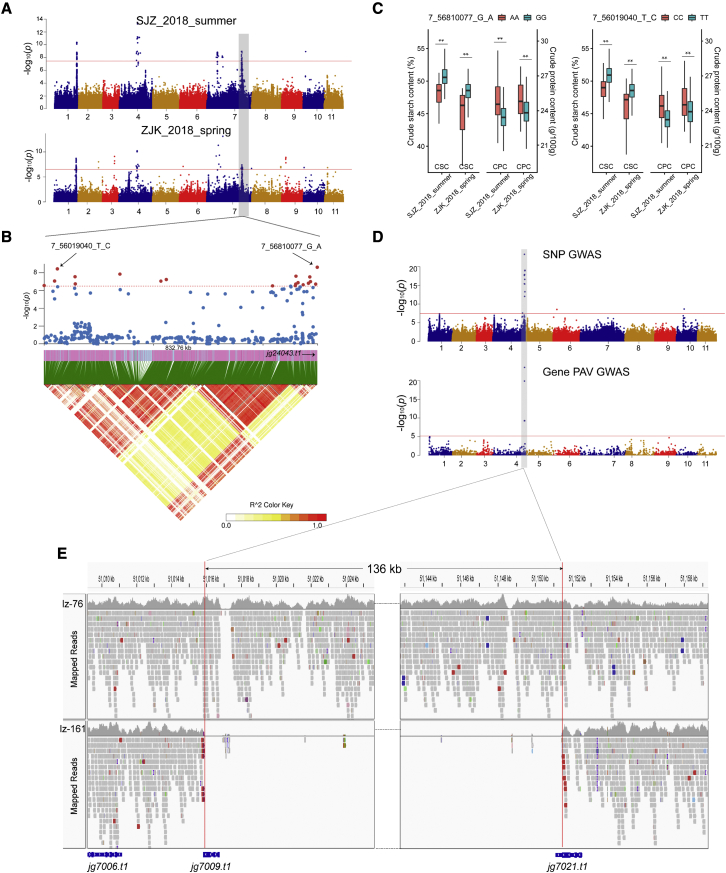
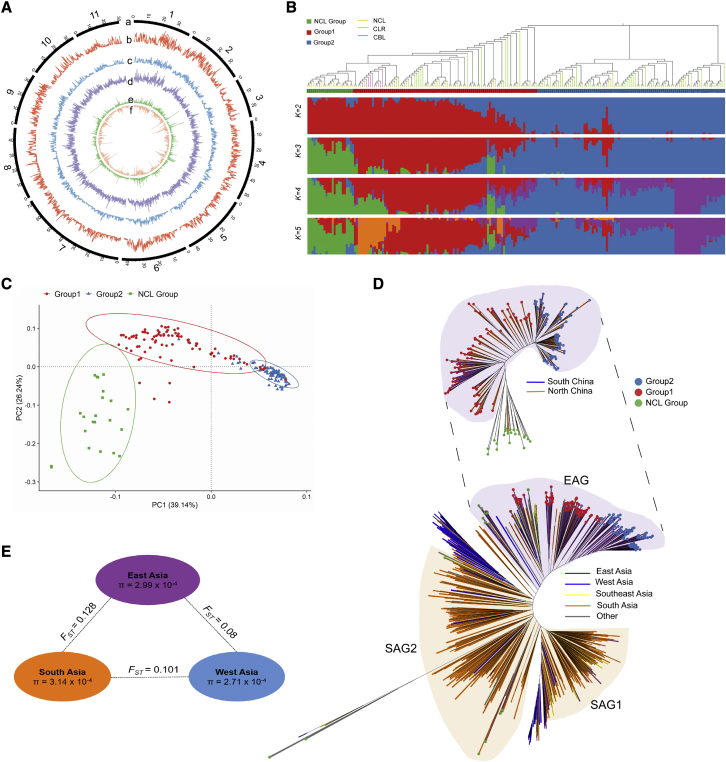
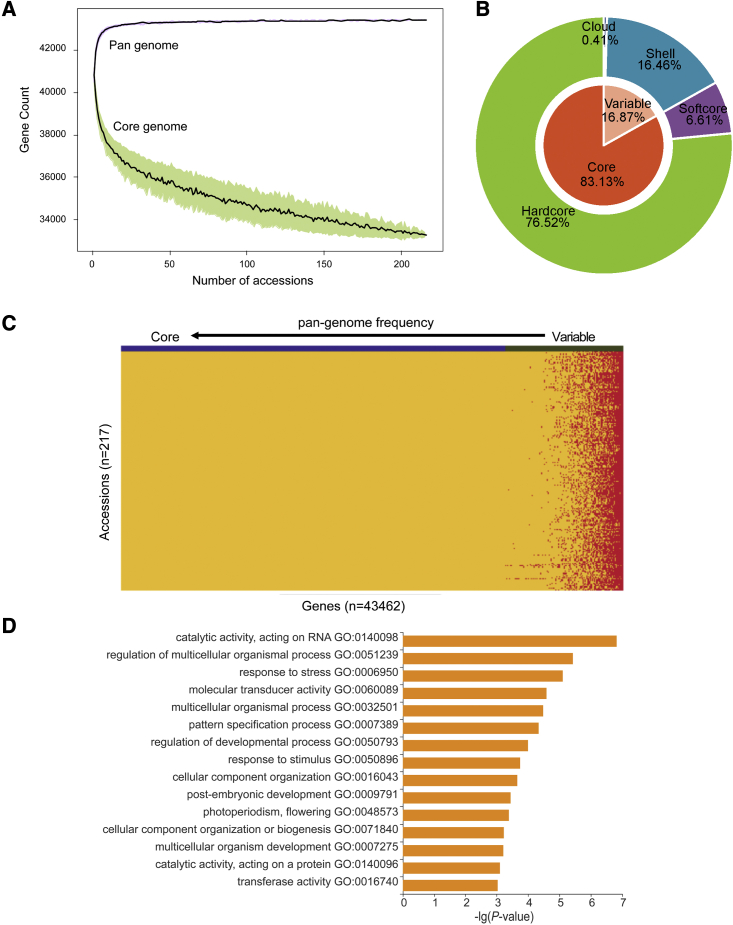
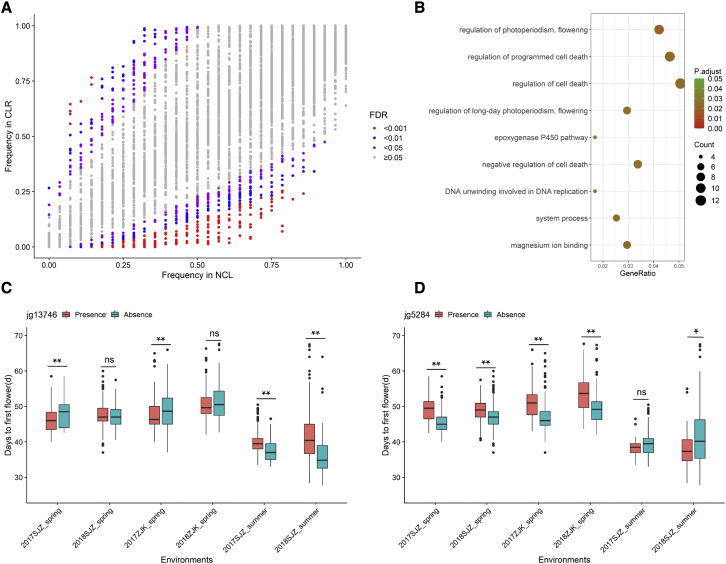
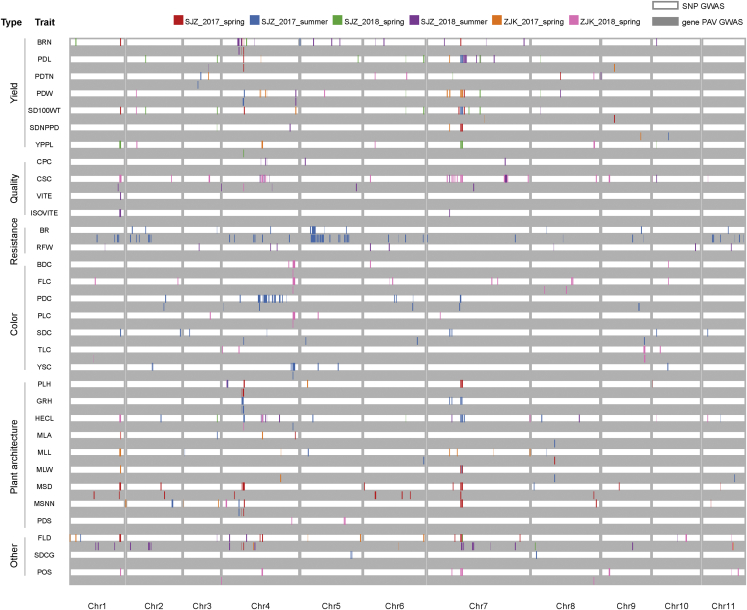
Mung bean is an economically important legume crop species that is used as a food, consumed as a vegetable, and used as an ingredient and even as a medicine. To explore the genomic diversity of mung bean, we assembled a high-quality reference genome (Vrad_JL7) that was ∼479.35 Mb in size, with a contig N50 length of 10.34 Mb. A total of 40,125 protein-coding genes were annotated, representing ∼96.9% of the genetic region. We also sequenced 217 accessions, mainly landraces and cultivars from China, and identified 2,229,343 high-quality single-nucleotide polymorphisms (SNPs). Population structure revealed that the Chinese accessions diverged into two groups and were distinct from non-Chinese lines. Genetic diversity analysis based on genomic data from 750 accessions in 23 countries supported the hypothesis that mung bean was first domesticated in south Asia and introduced to east Asia probably through the Silk Road. We constructed the first pan-genome of mung bean germplasm and assembled 287.73 Mb of non-reference sequences. Among the genes, 83.1% were core genes and 16.9% were variable. Presence/absence variation (PAV) events of nine genes involved in the regulation of the photoperiodic flowering pathway were identified as being under selection during the adaptation process to promote early flowering in the spring. Genome-wide association studies (GWASs) revealed 2,912 SNPs and 259 gene PAV events associated with 33 agronomic traits, including a SNP in the coding region of the SWEET10 homolog (jg24043) involved in crude starch content and a PAV event in a large fragment containing 11 genes for color-related traits. This high-quality reference genome and pan-genome will provide insights into mung bean breeding.
绿豆是一种经济上重要的豆类作物,既可用作食品,也可作为蔬菜食用,还可用作配料,甚至可用作药材。为了探索绿豆的基因组多样性,我们组装了一个高质量的参考基因组(Vrad_JL7),大小约为 479.35Mb,串联群 N50 长度为 10.34Mb。共注释了 40125 个蛋白质编码基因,约占遗传区域的 96.9%。我们还对 217 个样本进行了测序,这些样本主要来自中国的地方品种和栽培品种,共鉴定出 2229343 个高质量的单核苷酸多态性(SNP)。群体结构分析表明,中国样本分为两组,与非中国样本明显不同。基于来自 23 个国家的 750 个样本的基因组数据进行的遗传多样性分析支持了这样一种假说,即绿豆最初在南亚被驯化,并可能通过丝绸之路传入东亚。我们构建了第一个绿豆种质的泛基因组,组装了 287.73Mb 的非参考序列。在这些基因中,83.1%是核心基因,16.9%是可变基因。鉴定出参与光周期开花途径调控的 9 个基因的存在/缺失变异(PAV)事件,这些事件在适应过程中受到选择,以促进春季的早期开花。全基因组关联研究(GWASs)揭示了 2912 个 SNP 和 259 个基因 PAV 事件与 33 个农艺性状相关,包括编码区 SWEET10 同源物(jg24043)的一个 SNP,该 SNP与粗淀粉含量有关,以及一个包含 11 个与颜色相关性状的基因的大片段的 PAV 事件。这个高质量的参考基因组和泛基因组将为绿豆的育种提供新的见解。