Wang Yihang, Parmar Shaifaly, Schneekloth John S, Tiwary Pratyush
Biophysics Program and Institute for Physical Science and Technology, University of Maryland, College Park, Maryland 20742, United States.
Chemical Biology Laboratory, Center for Cancer Research, National Cancer Institute, Frederick, Maryland 21702, United States.
ACS Cent Sci. 2022 Jun 22;8(6):741-748. doi: 10.1021/acscentsci.2c00149. Epub 2022 May 16.
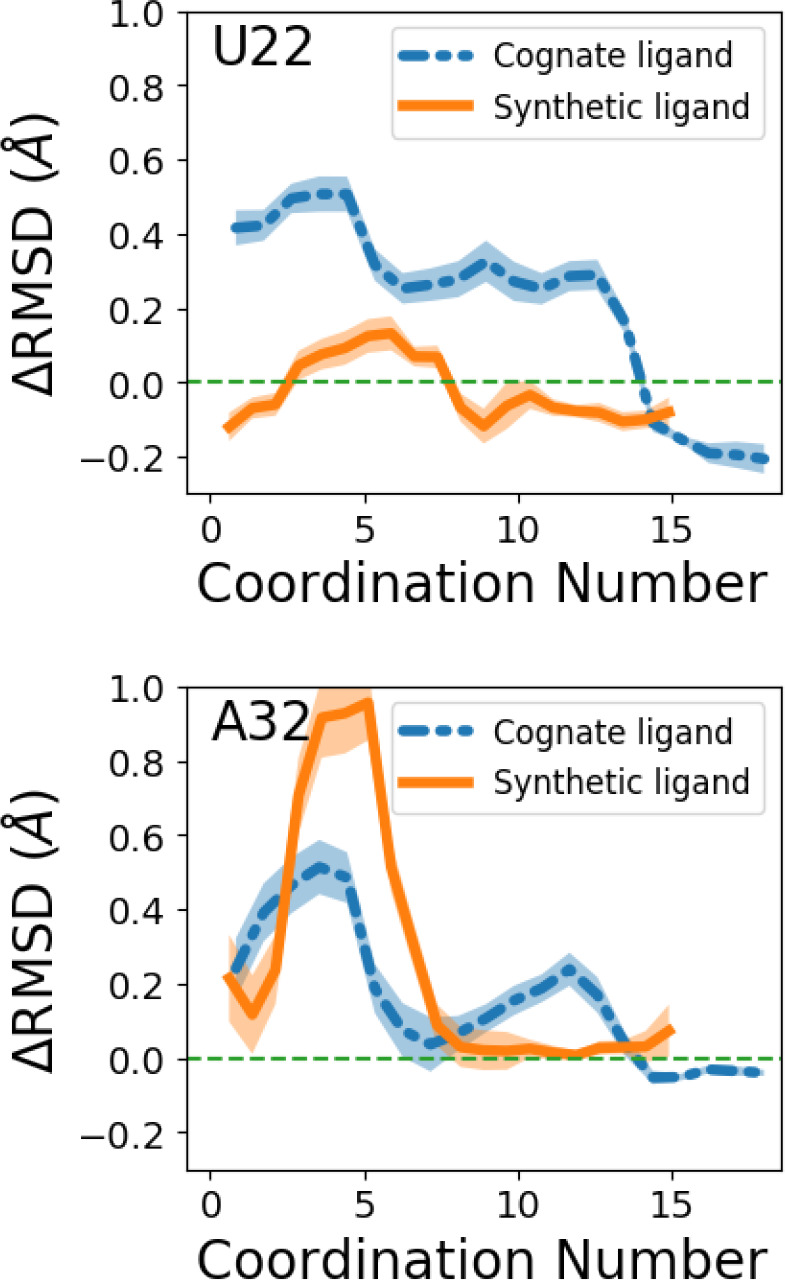
While there is increasing interest in the study of RNA as a therapeutic target, efforts to understand RNA-ligand recognition at the molecular level lag far behind our understanding of protein-ligand recognition. This problem is complicated due to the more than 10 orders of magnitude in time scales involved in RNA dynamics and ligand binding events, making it not straightforward to design experiments or simulations. Here, we make use of artificial intelligence (AI)-augmented molecular dynamics simulations to directly observe ligand dissociation for cognate and synthetic ligands from a riboswitch system. The site-specific flexibility profiles from our simulations are compared with measurements of flexibility using selective 2' hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP). Our simulations reproduce known relative binding affinity profiles for the cognate and synthetic ligands, and pinpoint how both ligands make use of different aspects of riboswitch flexibility. On the basis of our dissociation trajectories, we also make and validate predictions of pairs of mutations for both the ligand systems that would show differing binding affinities. These mutations are distal to the binding site and could not have been predicted solely on the basis of structure. The methodology demonstrated here shows how molecular dynamics simulations with all-atom force-fields have now come of age in making predictions that complement existing experimental techniques and illuminate aspects of systems otherwise not trivial to understand.
虽然将RNA作为治疗靶点的研究越来越受到关注,但在分子水平上理解RNA-配体识别的努力远远落后于我们对蛋白质-配体识别的理解。由于RNA动力学和配体结合事件涉及的时间尺度相差超过10个数量级,这个问题变得复杂起来,这使得设计实验或模拟并非易事。在这里,我们利用人工智能增强的分子动力学模拟直接观察来自核糖开关系统的同源和合成配体的配体解离。我们模拟得到的位点特异性灵活性概况与使用引物延伸和突变分析的选择性2'-羟基酰化(SHAPE-MaP)对灵活性的测量结果进行了比较。我们的模拟重现了同源和合成配体已知的相对结合亲和力概况,并指出了两种配体如何利用核糖开关灵活性的不同方面。基于我们的解离轨迹,我们还对两种配体系统中显示不同结合亲和力的突变对进行了预测并验证。这些突变位于结合位点的远端,不能仅根据结构进行预测。这里展示的方法表明,使用全原子力场的分子动力学模拟现在已经成熟,可以做出补充现有实验技术的预测,并阐明系统中其他难以理解的方面。