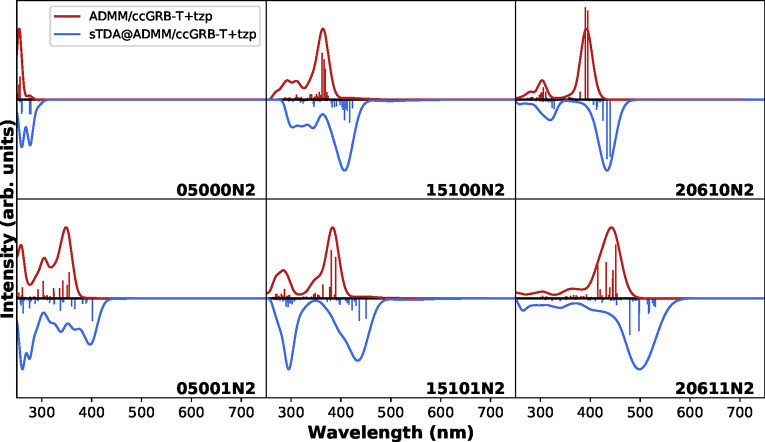
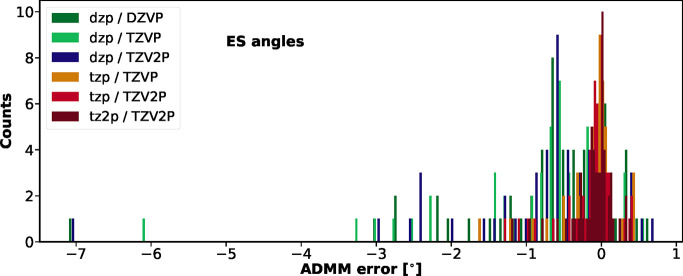
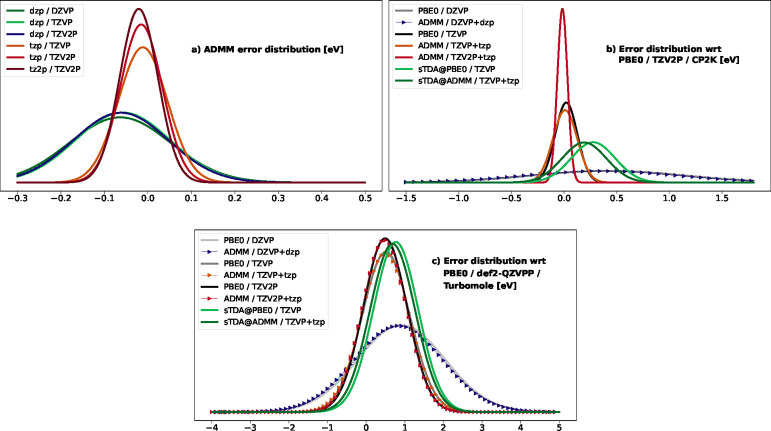
Hehn Anna-Sophia, Sertcan Beliz, Belleflamme Fabian, Chulkov Sergey K, Watkins Matthew B, Hutter Jürg
Department of Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland.
School of Mathematics and Physics, University of Lincoln, Brayford Pool, Lincoln LN67TS, United Kingdom.
J Chem Theory Comput. 2022 Jul 12;18(7):4186-4202. doi: 10.1021/acs.jctc.2c00144. Epub 2022 Jun 27.
Time-dependent density functional theory has become state-of-the-art for describing photophysical and photochemical processes in extended materials because of its affordable cost. The inclusion of exact exchange was shown to be essential for the correct description of the long-range asymptotics of electronic interactions and thus a well-balanced description of valence, Rydberg, and charge-transfer excitations. Several approaches for an efficient treatment of exact exchange have been established for the ground state, while implementations for excited-state properties are rare. Furthermore, the high computational costs required for excited-state properties in comparison to ground-state computations often hinder large-scale applications on periodic systems with hybrid functional accuracy. We therefore propose two approximate schemes for improving computational efficiency for the treatment of exact exchange. Within the auxiliary density matrix method (ADMM), exact exchange is estimated using a relatively small auxiliary basis and the introduced basis set incompleteness error is compensated by an exchange density functional correction term. Benchmark results for a test set of 35 molecules demonstrate that the mean absolute error introduced by ADMM is smaller than 0.3 pm for excited-state bond lengths and in the range of 0.02-0.04 eV for vertical excitation, adiabatic excitation, and fluorescence energies. Computational timings for a series of covalent-organic frameworks demonstrate that a speed-up of at least 1 order of magnitude can be achieved for excited-state geometry optimizations in comparison to conventional hybrid functionals. The second method is to use a semiempirical tight binding approximation for both Coulomb and exchange contributions to the excited-state kernel. This simplified Tamm-Dancoff approximation (sTDA) achieves an accuracy comparable to approximated hybrid density functional theory when referring to highly accurate coupled-cluster reference data. We find that excited-state bond lengths deviate by 1.1 pm on average and mean absolute errors in vertical excitation, adiabatic excitation, and fluorescence energies are in the range of 0.2-0.5 eV. In comparison to ADMM-approximated hybrid functional theory, sTDA accelerates the computation of broad-band excitation spectra by 1 order of magnitude, suggesting its potential use for large-scale screening purposes.
由于成本可承受,含时密度泛函理论已成为描述扩展材料中光物理和光化学过程的前沿方法。已表明,包含精确交换对于正确描述电子相互作用的长程渐近性至关重要,因此对价态、里德堡态和电荷转移激发进行了平衡良好的描述。对于基态,已经建立了几种有效处理精确交换的方法,而激发态性质的实现则很少见。此外,与基态计算相比,激发态性质所需的高计算成本常常阻碍具有杂化泛函精度的周期性系统的大规模应用。因此,我们提出了两种近似方案来提高处理精确交换的计算效率。在辅助密度矩阵方法(ADMM)中,使用相对较小的辅助基来估计精确交换,并通过交换密度泛函校正项来补偿引入的基组不完备误差。对一组35个分子的测试集的基准结果表明,ADMM引入的平均绝对误差对于激发态键长小于0.3皮米,对于垂直激发、绝热激发和荧光能量在0.02 - 0.04电子伏特范围内。一系列共价有机框架的计算时间表明,与传统杂化泛函相比,激发态几何优化可实现至少1个数量级的加速。第二种方法是对激发态核的库仑和交换贡献都使用半经验紧束缚近似。这种简化的Tamm - Dancoff近似(sTDA)在参考高精度耦合簇参考数据时,实现了与近似杂化密度泛函理论相当的精度。我们发现激发态键长平均偏差为1.1皮米,垂直激发、绝热激发和荧光能量的平均绝对误差在0.2 - 0.5电子伏特范围内。与ADMM近似的杂化泛函理论相比,sTDA将宽带激发光谱的计算加速了1个数量级,表明其在大规模筛选方面的潜在用途。