Stark Neurosciences Research Institute, Indiana University-School of Medicine, Indianapolis, IN, USA.
Department of Medical and Molecular Genetics, Indiana University-School of Medicine, Indianapolis, IN, USA.
Mol Neurodegener. 2022 Jun 28;17(1):47. doi: 10.1186/s13024-022-00545-9.
Despite its identification as a key checkpoint regulator of microglial activation in Alzheimer's disease, the overarching role of CX3CR1 signaling in modulating mechanisms of Aβ driven neurodegeneration, including accumulation of hyperphosphorylated tau is not well understood.
Accumulation of soluble and insoluble Aβ species, microglial activation, synaptic dysregulation, and neurodegeneration is investigated in 4- and 6-month old 5xFAD;Cx3cr1 and 5xFAD;Cx3cr1 mice using immunohistochemistry, western blotting, transcriptomic and quantitative real time PCR analyses of purified microglia. Flow cytometry based, in-vivo Aβ uptake assays are used for characterization of the effects of CX3CR1-signaling on microglial phagocytosis and lysosomal acidification as indicators of clearance of methoxy-X-04 fibrillar Aβ. Lastly, we use Y-maze testing to analyze the effects of Cx3cr1 deficiency on working memory.
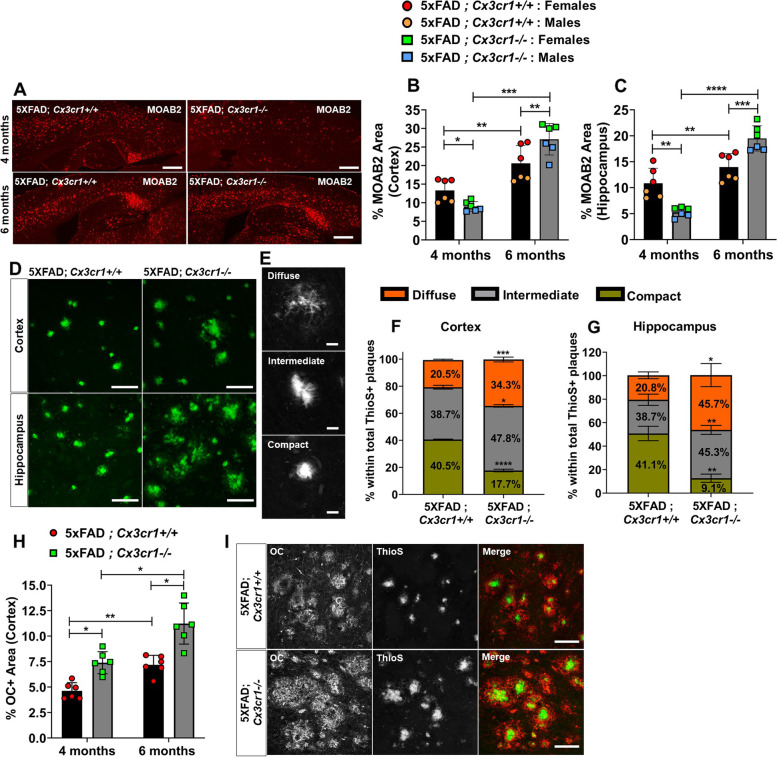
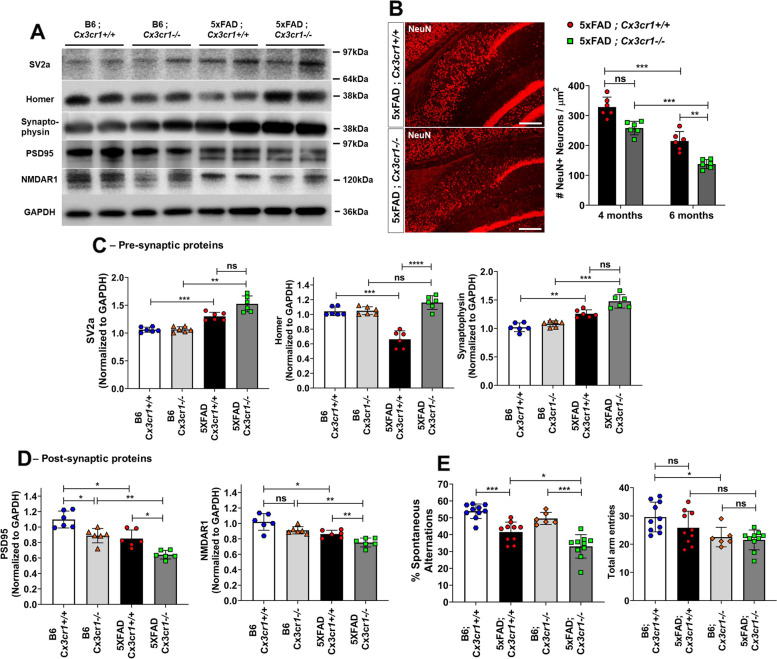
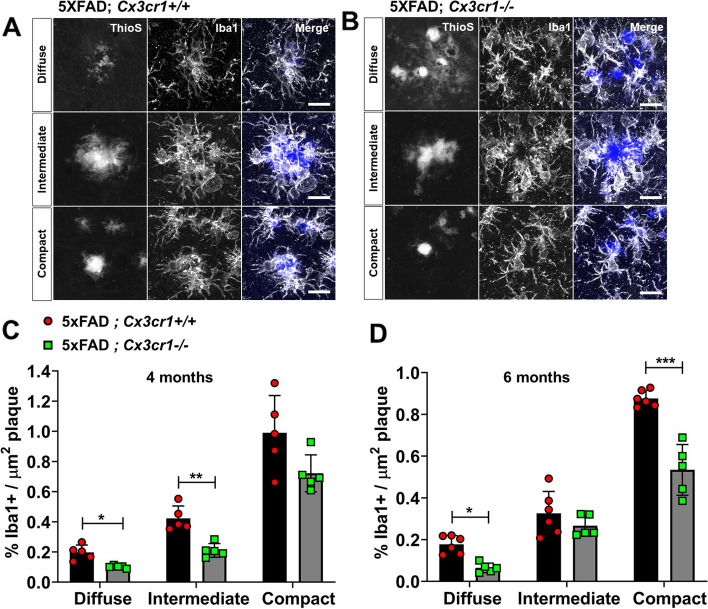
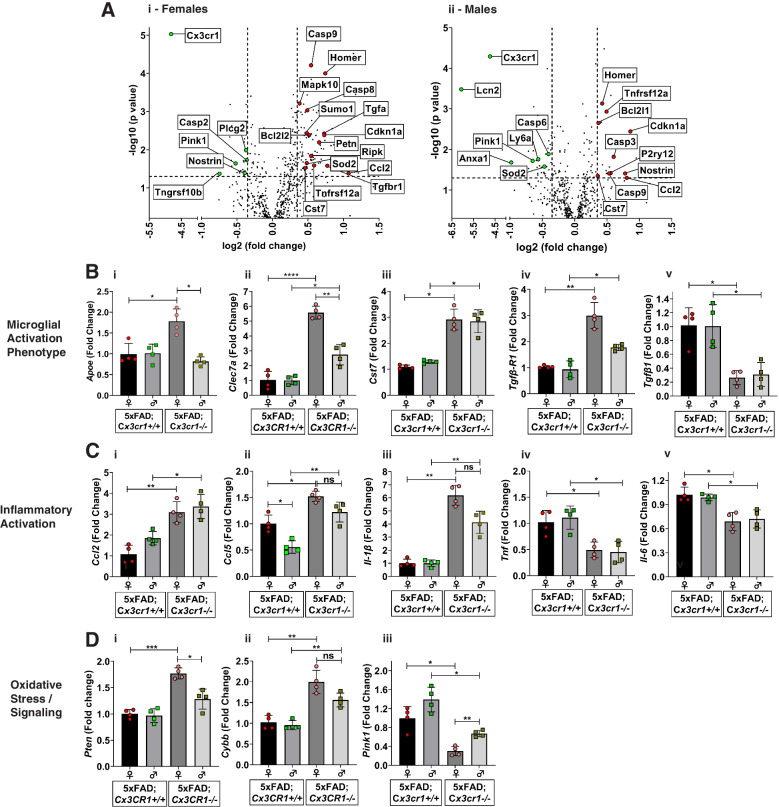
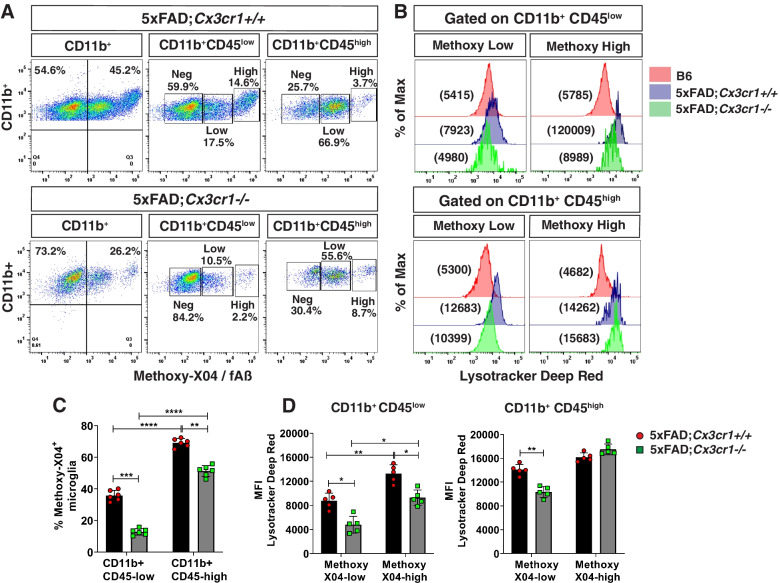
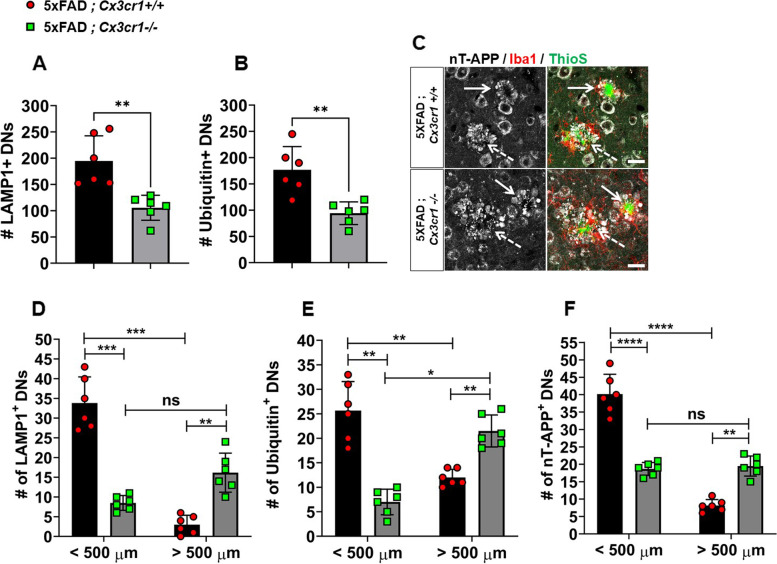
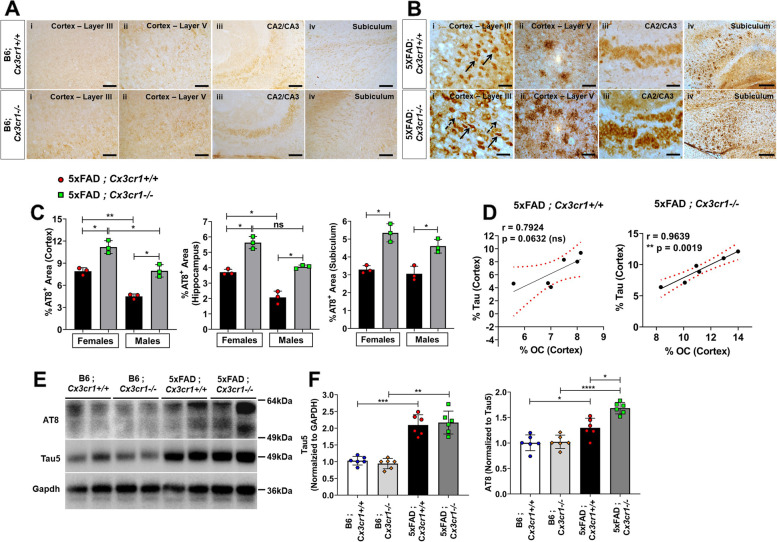
Disease progression in 5xFAD;Cx3cr1 mice is characterized by increased deposition of filamentous plaques that display defective microglial plaque engagement. Microglial Aβ phagocytosis and lysosomal acidification in 5xFAD;Cx3cr1 mice is impaired in-vivo. Interestingly, Cx3cr1 deficiency results in heighted accumulation of neurotoxic, oligomeric Aβ, along with severe neuritic dystrophy, preferential loss of post-synaptic densities, exacerbated tau pathology, neuronal loss and cognitive impairment. Transcriptomic analyses using cortical RNA, coupled with qRT-PCR using purified microglia from 6 month-old mice indicate dysregulated TGFβ-signaling and heightened ROS metabolism in 5xFAD;Cx3cr1 mice. Lastly, microglia in 6 month-old 5xFAD;Cx3cr1 mice express a 'degenerative' phenotype characterized by increased levels of Ccl2, Ccl5, Il-1β, Pten and Cybb along with reduced Tnf, Il-6 and Tgfβ1 mRNA.
Cx3cr1 deficiency impairs microglial uptake and degradation of fibrillar Aβ, thereby triggering increased accumulation of neurotoxic Aβ species. Furthermore, loss of Cx3cr1 results in microglial dysfunction typified by dampened TGFβ-signaling, increased oxidative stress responses and dysregulated pro-inflammatory activation. Our results indicate that Aβ-driven microglial dysfunction in Cx3cr1 mice aggravates tau hyperphosphorylation, neurodegeneration, synaptic dysregulation and impairs working memory.
尽管 CX3CR1 信号在调节阿尔茨海默病中微胶质细胞激活的关键检查点调节剂方面已被确定,但 CX3CR1 信号在调节包括过度磷酸化 tau 在内的 Aβ 驱动的神经退行性变机制方面的总体作用尚不清楚。
使用免疫组织化学、western blot、纯化的小胶质细胞的转录组和定量实时 PCR 分析,研究了 4 个月和 6 个月大的 5xFAD;Cx3cr1 和 5xFAD;Cx3cr1 小鼠中可溶性和不溶性 Aβ 物种的积累、小胶质细胞激活、突触失调和神经退行性变。使用基于流式细胞术的体内 Aβ 摄取测定法来表征 CX3CR1 信号对小胶质细胞吞噬作用和溶酶体酸化的影响,作为甲氧基-X-04 纤维状 Aβ清除的指标。最后,我们使用 Y 迷宫测试分析 Cx3cr1 缺乏对工作记忆的影响。
5xFAD;Cx3cr1 小鼠的疾病进展特征为丝状斑块的沉积增加,这些斑块显示出微胶质斑块结合的缺陷。5xFAD;Cx3cr1 小鼠中的小胶质细胞 Aβ 吞噬作用和溶酶体酸化在体内受损。有趣的是,Cx3cr1 缺乏导致神经毒性寡聚体 Aβ 的积累增加,以及严重的神经突营养不良、突触后密度的优先丧失、tau 病理学的加剧、神经元丧失和认知障碍。使用皮质 RNA 进行的转录组分析,以及使用来自 6 个月大小鼠的纯化小胶质细胞进行的 qRT-PCR 表明,5xFAD;Cx3cr1 小鼠中的 TGFβ 信号和 ROS 代谢失调。最后,6 个月大的 5xFAD;Cx3cr1 小鼠的小胶质细胞表达一种“退行性”表型,其特征是 Ccl2、Ccl5、Il-1β、Pten 和 Cybb 的水平升高,而 Tnf、Il-6 和 Tgfβ1mRNA 的水平降低。
Cx3cr1 缺乏会损害小胶质细胞对纤维状 Aβ 的摄取和降解,从而导致神经毒性 Aβ 物种的积累增加。此外,Cx3cr1 的缺失导致小胶质细胞功能障碍,表现为 TGFβ 信号减弱、氧化应激反应增加和促炎激活失调。我们的结果表明,Cx3cr1 小鼠中的 Aβ 驱动的小胶质细胞功能障碍加剧了 tau 过度磷酸化、神经退行性变、突触失调和工作记忆受损。