Department of General Surgery, Shengjing Hospital of China Medical University, 36 Sanhao Street, Heping District, Shenyang 110004, China.
Biomed Res Int. 2022 Jun 21;2022:1893351. doi: 10.1155/2022/1893351. eCollection 2022.
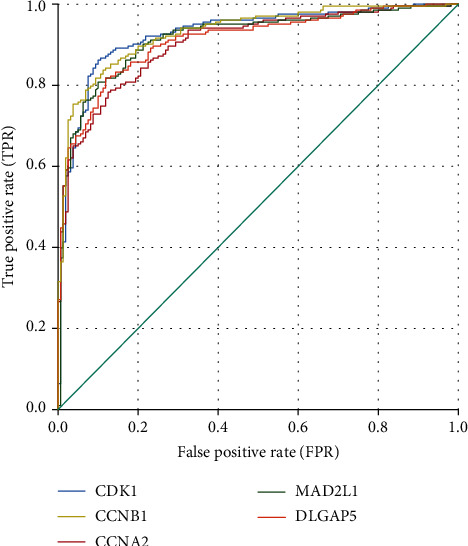
Colon adenocarcinoma (COAD) is among the most common digestive system malignancies worldwide, and its pathogenesis and gene signatures remain unclear. This study explored the genetic characteristics and molecular mechanisms underlying colon cancer development. Three gene expression data sets were obtained from the Gene Expression Omnibus (GEO) database. GEO2R was used to determine differentially expressed genes (DEGs) between COAD and normal tissues. Then, the intersection of the data sets was obtained. Metascape was used to perform the functional enrichment analyses. Next, STRING was used to build protein-protein interaction (PPI) networks. Hub genes were identified and analysed using Cytoscape. Next, survival analysis and expression analysis of the hub genes were performed. ROC curve analysis was performed for further test of the diagnostic efficacy. Finally, alterations in the hub genes were predicted and analysed by cBioPortal. Altogether, 436 DEGs were detected. The DEGs were mainly enriched in cell cycle phase transition, nuclear division, meiotic nuclear division, and cytokinesis. Based on PPI networks, 20 hub genes were selected. Among them, 6 hub genes (, , , , , and ) showed significant prognostic value in colon cancer ( < 0.05), while 5 hub genes (, , , , and ) were associated with early colon cancer diagnosis and ROC curve analysis showed good diagnostic accuracy. In conclusion, integrated bioinformatics analysis was used to identify hub genes that reveal the potential mechanism of carcinogenesis and progression of colon cancer. The hub genes might be novel biomarkers for early diagnosis, treatment, and prognosis of colon cancer.
结肠腺癌(COAD)是全球最常见的消化系统恶性肿瘤之一,其发病机制和基因特征尚不清楚。本研究探讨了结肠癌发生的遗传特征和分子机制。从基因表达综合数据库(GEO)中获取了三个基因表达数据集。使用 GEO2R 确定 COAD 和正常组织之间的差异表达基因(DEG)。然后,获取数据集的交集。使用 Metascape 进行功能富集分析。接下来,使用 STRING 构建蛋白质-蛋白质相互作用(PPI)网络。使用 Cytoscape 识别和分析枢纽基因。接下来,对枢纽基因进行生存分析和表达分析。进行 ROC 曲线分析以进一步测试诊断效能。最后,通过 cBioPortal 预测和分析枢纽基因的改变。共检测到 436 个 DEG。DEG 主要富集在细胞周期相转变、核分裂、减数分裂核分裂和胞质分裂。基于 PPI 网络,选择了 20 个枢纽基因。其中,6 个枢纽基因(、、、、和)在结肠癌中具有显著的预后价值(<0.05),而 5 个枢纽基因(、、、、和)与早期结肠癌的诊断相关,ROC 曲线分析显示出良好的诊断准确性。总之,通过综合生物信息学分析,确定了枢纽基因,揭示了结肠癌发生和进展的潜在机制。这些枢纽基因可能是结肠癌早期诊断、治疗和预后的新的生物标志物。