Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Prague 6, Czech Republic.
Faculty of Natural Sciences, Charles University, Praha 2, Czech Republic.
Proteins. 2022 Dec;90(12):2067-2079. doi: 10.1002/prot.26398. Epub 2022 Jul 27.
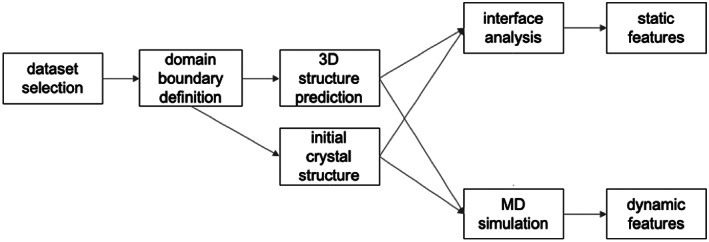
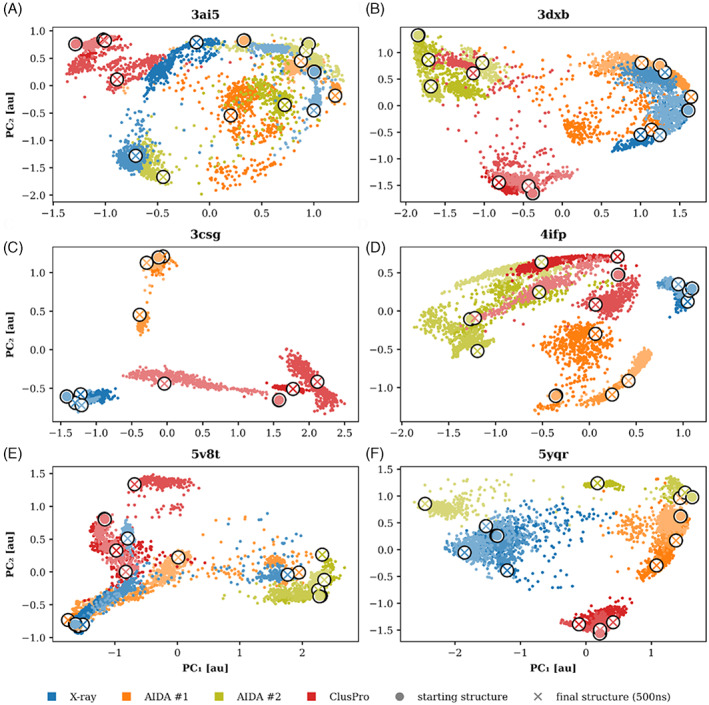
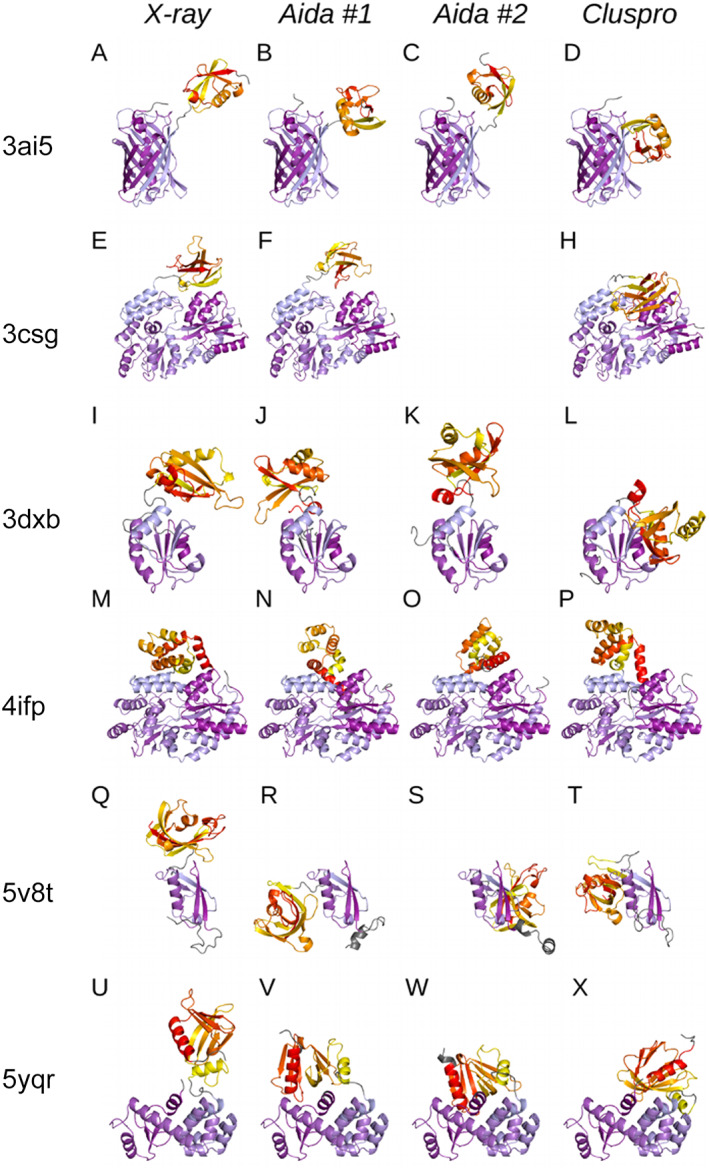
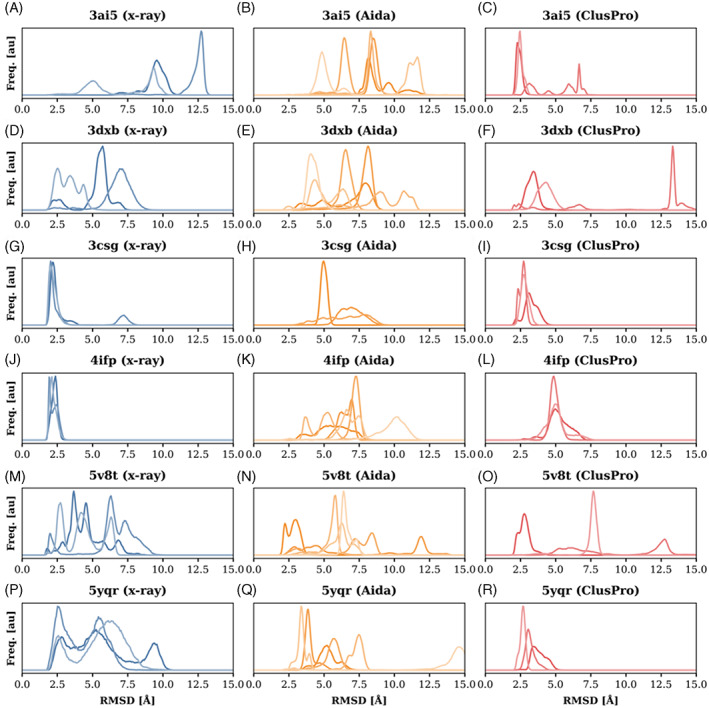
Proteins are naturally formed by domains edging their functional and structural properties. A domain out of the context of an entire protein can retain its structure and to some extent also function on its own. These properties rationalize construction of artificial fusion multidomain proteins with unique combination of various functions. Information on the specific functional and structural characteristics of individual domains in the context of new artificial fusion proteins is inevitably encoded in sequential order of composing domains defining their mutual spatial positions. So the challenges in designing new proteins with new domain combinations lie dominantly in structure/function prediction and its context dependency. Despite the enormous body of publications on artificial fusion proteins, the task of their structure/function prediction is complex and nontrivial. The degree of spatial freedom facilitated by a linker between domains and their mutual orientation driven by noncovalent interactions is beyond a simple and straightforward methodology to predict their structure with reasonable accuracy. In the presented manuscript, we tested methodology using available modeling tools and computational methods. We show that the process and methodology of such prediction are not straightforward and must be done with care even when recently introduced AlphaFold II is used. We also addressed a question of benchmarking standards for prediction of multidomain protein structures-x-ray or Nuclear Magnetic Resonance experiments. On the study of six two-domain protein chimeras as well as their composing domains and their x-ray structures selected from PDB, we conclude that the major obstacle for justified prediction is inappropriate sampling of the conformational space by the explored methods. On the other hands, we can still address particular steps of the methodology and improve the process of chimera proteins prediction.
蛋白质是通过边缘其功能和结构特性的结构域自然形成的。结构域在整个蛋白质的背景之外,可以保留其结构,并且在某种程度上也可以独立发挥其功能。这些特性为构建具有独特功能组合的人工融合多结构域蛋白质提供了合理性。关于新人工融合蛋白质中各个结构域的特定功能和结构特性的信息不可避免地编码在构成结构域的顺序中,定义了它们的相互空间位置。因此,设计具有新结构域组合的新型蛋白质的挑战主要在于结构/功能预测及其上下文依赖性。尽管有关人工融合蛋白质的出版物数量巨大,但它们的结构/功能预测任务仍然很复杂。结构域之间的连接体提供的空间自由度以及非共价相互作用驱动的它们的相互取向程度超出了使用简单直接的方法来合理准确地预测其结构的范围。在本手稿中,我们使用了可用的建模工具和计算方法来测试方法。我们表明,即使使用最近引入的 AlphaFold II,这种预测的过程和方法也不简单,必须谨慎进行。我们还讨论了多维蛋白质结构预测的基准标准问题-x 射线或核磁共振实验。通过对从 PDB 中选择的六个两结构域蛋白质嵌合体及其组成结构域和它们的 x 射线结构的研究,我们得出结论,合理预测的主要障碍是所探索的方法对构象空间的取样不当。另一方面,我们仍然可以解决方法的特定步骤,并改进嵌合体蛋白质预测的过程。