Matsuno Shumpei, Ohue Masahito, Akiyama Yutaka
Department of Computer Science, School of Computing, Tokyo Institute of Technology, Meguro-ku, Tokyo 152-8550, Japan.
AIST-TokyoTech Real World Big-Data Computation Open Innovation Laboratory (RWBC-OIL), National Institute of Advanced Industrial Science and Technology, Tsukuba, Ibaraki 305-8560, Japan.
Biophys Physicobiol. 2020 Feb 7;17:2-13. doi: 10.2142/biophysico.BSJ-2019050. eCollection 2020.
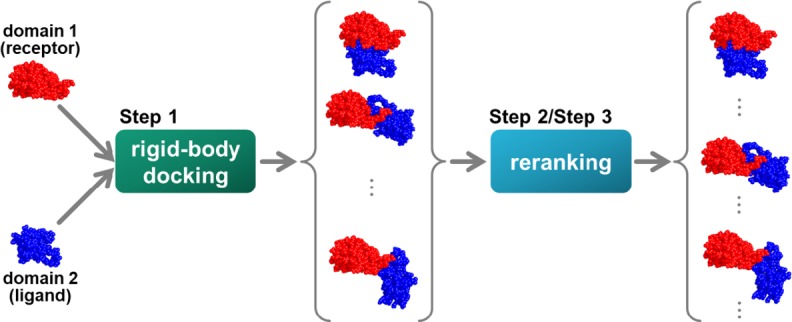
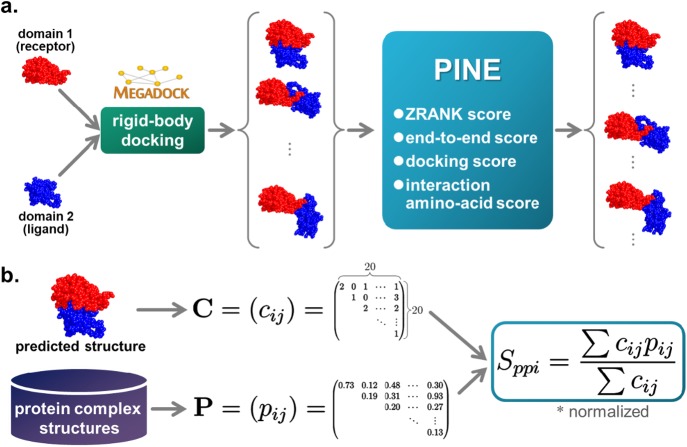
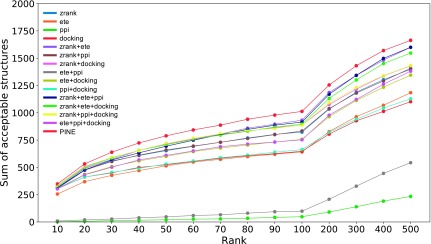
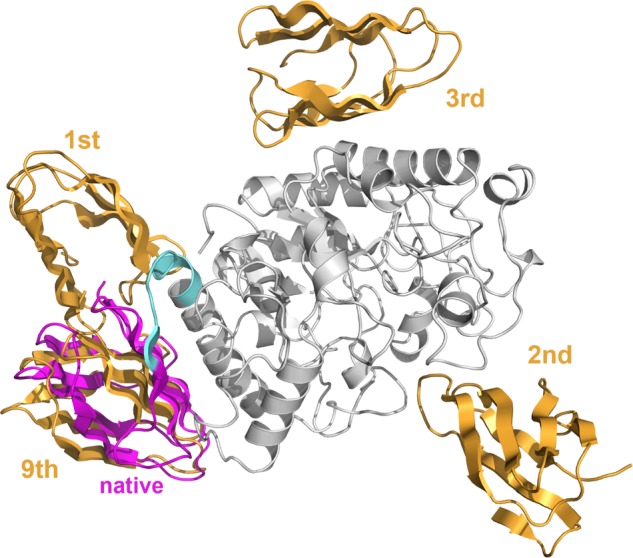
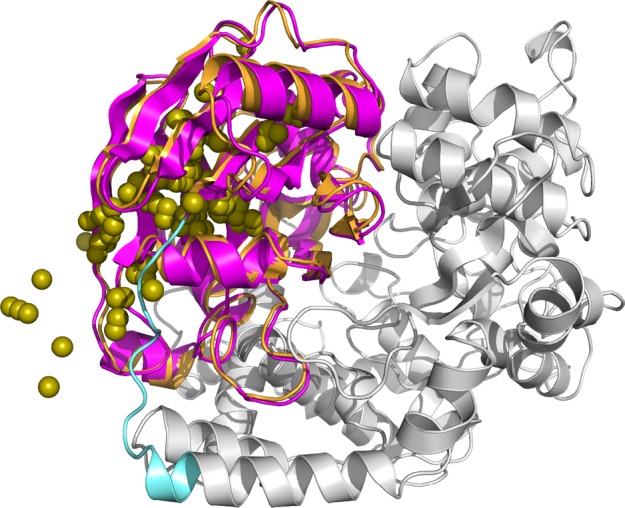
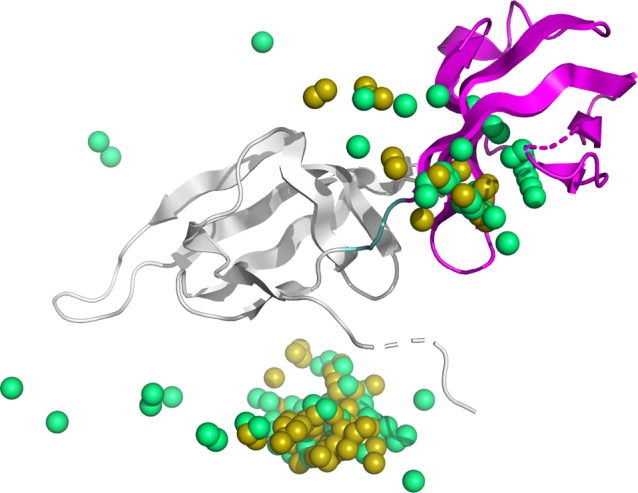
Protein functions can be predicted based on their three-dimensional structures. However, many multidomain proteins have unstable structures, making it difficult to determine the whole structure in biological experiments. Additionally, multidomain proteins are often decomposed and identified based on their domains, with the structure of each domain often found in public databases. Recent studies have advanced structure prediction methods of multidomain proteins through computational analysis. In existing methods, proteins that serve as templates are used for three-dimensional structure prediction. However, when no protein template is available, the accuracy of the prediction is decreased. This study was conducted to predict the structures of multidomain proteins without the need for whole structure templates. We improved structure prediction methods by performing rigid-body docking from the structure of each domain and reranking a structure closer to the correct structure to have a higher value. In the proposed method, the score for the domain-domain interaction obtained without a structural template of the multidomain protein and score for the three-dimensional structure obtained during docking calculation were newly incorporated into the score function. We successfully predicted the structures of 50 of 55 multidomain proteins examined in the test dataset. Interaction residue pair information of the protein-protein complex interface contributes to domain reorganizations even when a structural template for a multidomain protein cannot be obtained. This approach may be useful for predicting the structures of multidomain proteins with important biochemical functions.
蛋白质的功能可以根据其三维结构进行预测。然而,许多多结构域蛋白质具有不稳定的结构,这使得在生物学实验中难以确定其完整结构。此外,多结构域蛋白质通常基于其结构域进行分解和鉴定,每个结构域的结构经常可以在公共数据库中找到。最近的研究通过计算分析改进了多结构域蛋白质的结构预测方法。在现有方法中,用作模板的蛋白质用于三维结构预测。然而,当没有蛋白质模板可用时,预测的准确性会降低。本研究旨在无需完整结构模板即可预测多结构域蛋白质的结构。我们通过从每个结构域的结构进行刚体对接并重新排列更接近正确结构的结构以使其具有更高的值来改进结构预测方法。在所提出的方法中,在没有多结构域蛋白质结构模板的情况下获得的结构域 - 结构域相互作用得分以及对接计算期间获得的三维结构得分被新纳入得分函数。我们成功预测了测试数据集中55个多结构域蛋白质中的50个的结构。即使无法获得多结构域蛋白质的结构模板,蛋白质 - 蛋白质复合物界面的相互作用残基对信息也有助于结构域重组。这种方法可能有助于预测具有重要生化功能的多结构域蛋白质的结构。