St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065.
Division of Pediatric Allergy and Immunology, Necmettin Erbakan University, Meram Medical Faculty, Konya 42080, Turkey.
Proc Natl Acad Sci U S A. 2023 Nov 14;120(46):e2314225120. doi: 10.1073/pnas.2314225120. Epub 2023 Nov 6.
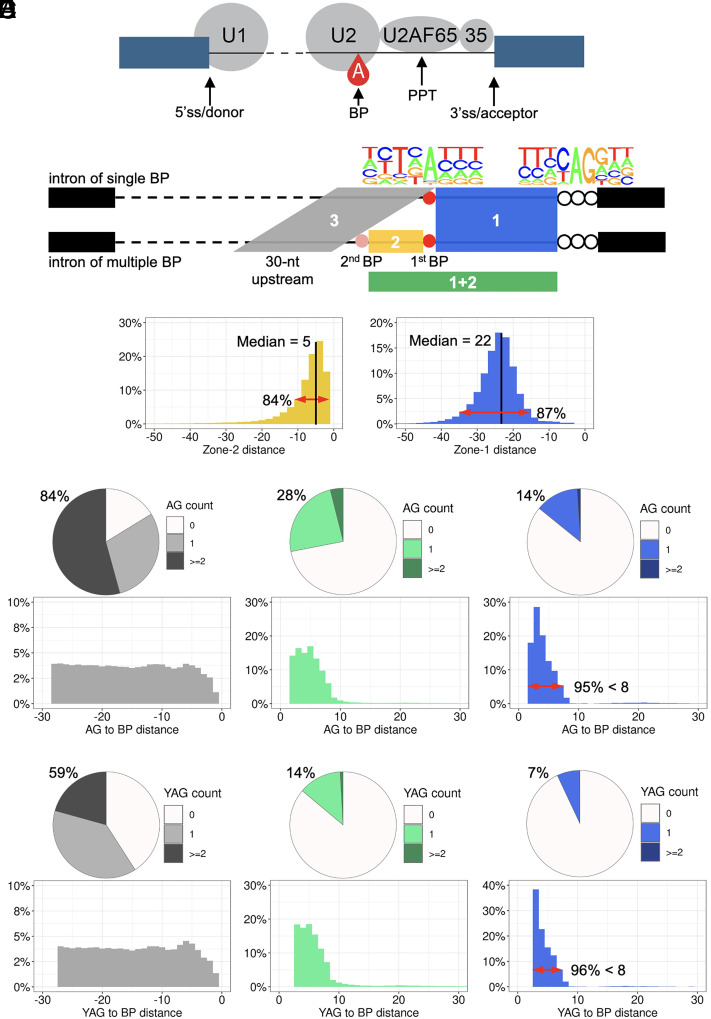
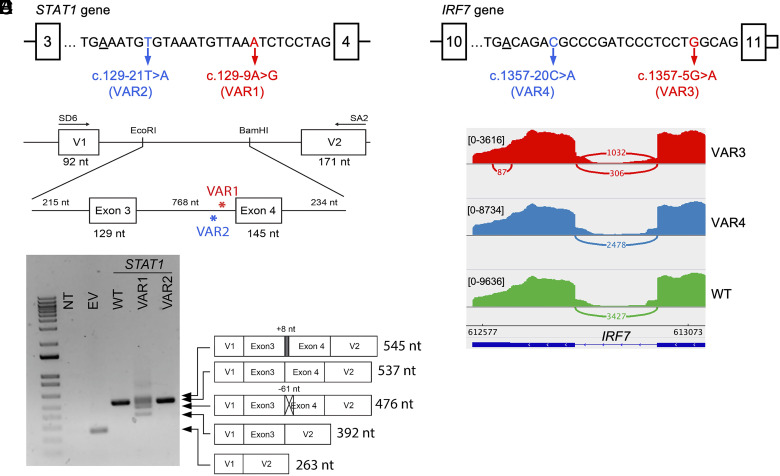
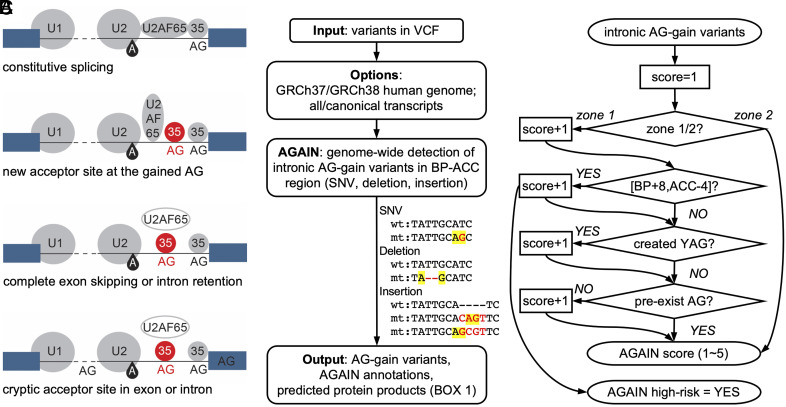
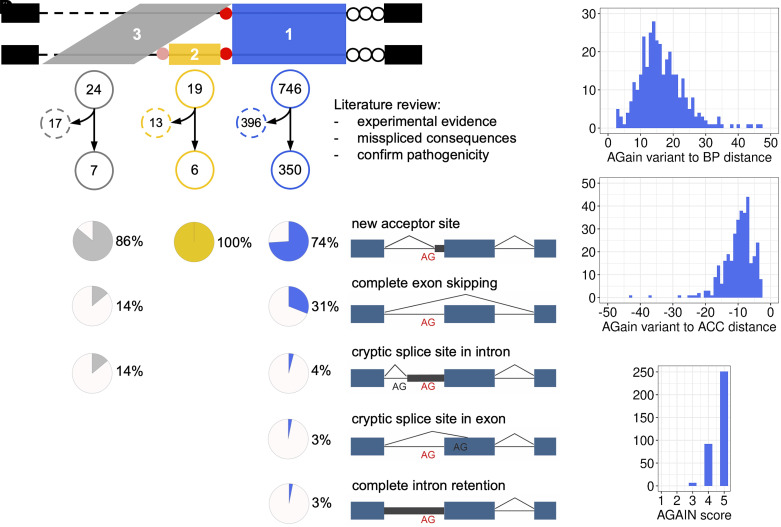
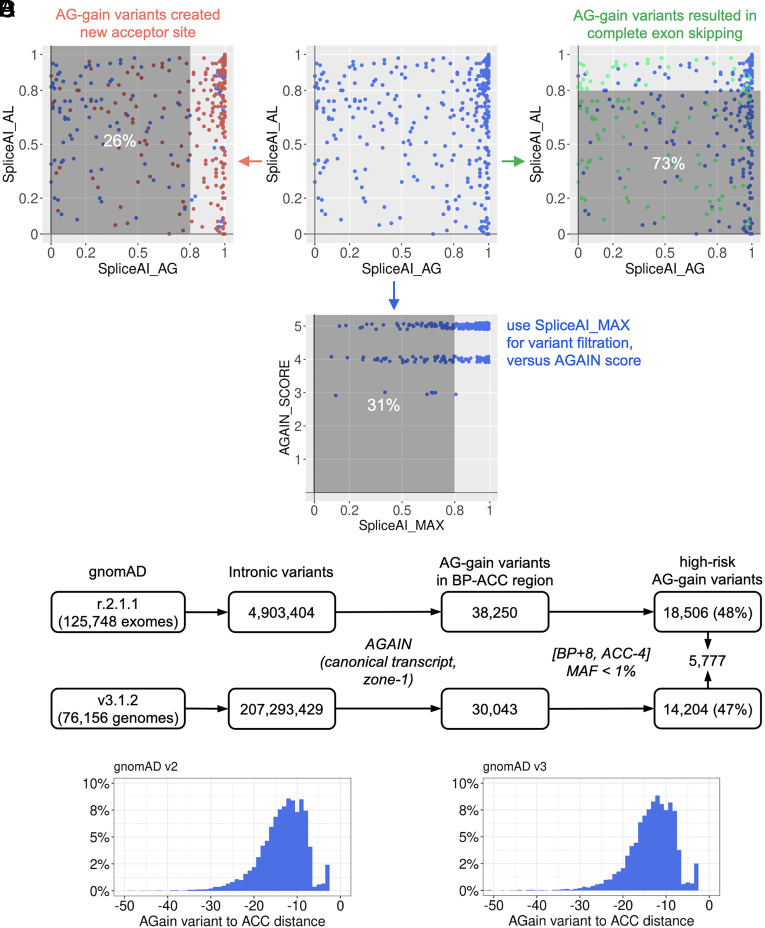
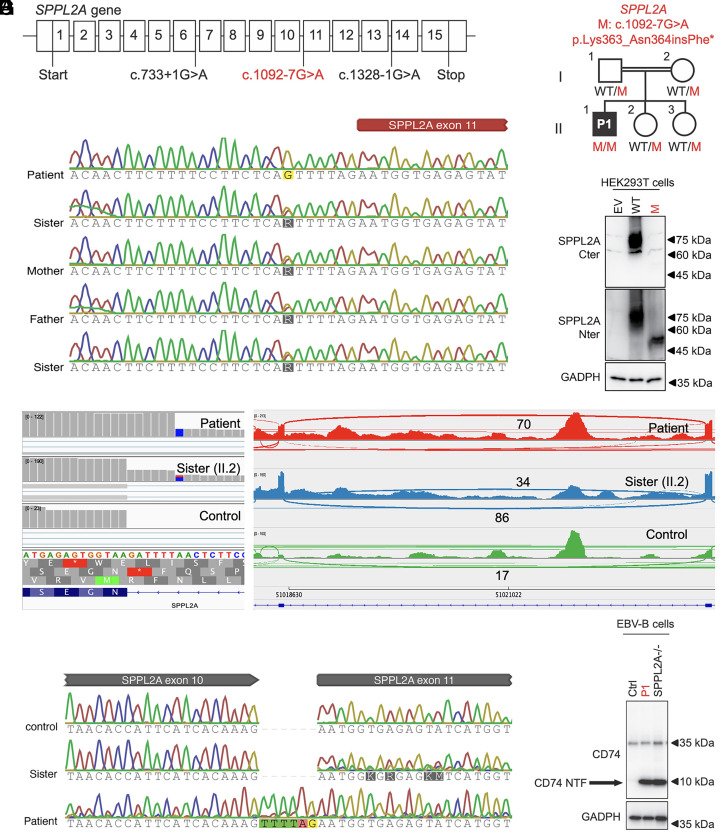
Human genetic variants that introduce an AG into the intronic region between the branchpoint (BP) and the canonical splice acceptor site (ACC) of protein-coding genes can disrupt pre-mRNA splicing. Using our genome-wide BP database, we delineated the BP-ACC segments of all human introns and found extreme depletion of AG/YAG in the [BP+8, ACC-4] high-risk region. We developed AGAIN as a genome-wide computational approach to systematically and precisely pinpoint intronic AG-gain variants within the BP-ACC regions. AGAIN identified 350 AG-gain variants from the Human Gene Mutation Database, all of which alter splicing and cause disease. Among them, 74% created new acceptor sites, whereas 31% resulted in complete exon skipping. AGAIN also predicts the protein-level products resulting from these two consequences. We performed AGAIN on our exome/genomes database of patients with severe infectious diseases but without known genetic etiology and identified a private homozygous intronic AG-gain variant in the antimycobacterial gene in a patient with mycobacterial disease. AGAIN also predicts a retention of six intronic nucleotides that encode an in-frame stop codon, turning AG-gain into stop-gain. This allele was then confirmed experimentally to lead to loss of function by disrupting splicing. We further showed that AG-gain variants inside the high-risk region led to misspliced products, while those outside the region did not, by two case studies in genes STAT1 and IRF7. We finally evaluated AGAIN on our 14 paired exome-RNAseq samples and found that 82% of AG-gain variants in high-risk regions showed evidence of missplicing. AGAIN is publicly available from https://hgidsoft.rockefeller.edu/AGAIN and https://github.com/casanova-lab/AGAIN.
人类基因组中,位于蛋白编码基因分支点(BP)和典型剪接受体位点(ACC)之间内含子区域的 AG 变异可导致前体 mRNA 剪接异常。我们利用全基因组 BP 数据库,描绘了所有人类内含子的 BP-ACC 区域,并发现 [BP+8,ACC-4]高风险区域中 AG/YAG 极端缺失。我们开发了 AGAIN 作为一种全基因组计算方法,用于系统、准确地确定 BP-ACC 区域内的内含子 AG 获得性变异。AGAING 从人类基因突变数据库中鉴定了 350 个 AG 获得性变异,这些变异均改变了剪接并导致疾病。其中,74%的变异创造了新的剪接受体位点,而 31%的变异导致外显子完全缺失。AGAING 还预测了这两种后果导致的蛋白质水平产物。我们在严重感染性疾病且无已知遗传病因的患者的外显子/基因组数据库中执行 AGAIN 分析,在一名患有分枝杆菌病的患者的抗分枝杆菌基因中发现了一个纯合的内含子 AG 获得性变异。AGAING 还预测了 6 个内含子核苷酸的保留,这些核苷酸编码一个无义的终止密码子,使 AG 获得性变异变成终止获得性变异。该等位基因随后通过实验证实可通过破坏剪接导致功能丧失。我们进一步通过 STAT1 和 IRF7 两个基因的两个案例研究表明,高风险区域内的 AG 获得性变异会导致剪接异常,而区域外的变异则不会。我们最后在我们的 14 对外显子 RNA-seq 样本中评估 AGAIN,发现高风险区域内的 82%的 AG 获得性变异有剪接异常的证据。AGAING 可在 https://hgidsoft.rockefeller.edu/AGAIN 和 https://github.com/casanova-lab/AGAIN 上公开获取。