Food and Nutrition Program, Kuwait Institute for Scientific Research, Safat 13109, Kuwait.
Systems and Software Development Department, Kuwait Institute for Scientific Research, Safat 13109, Kuwait.
Bioinformatics. 2022 Sep 2;38(17):4088-4099. doi: 10.1093/bioinformatics/btac519.
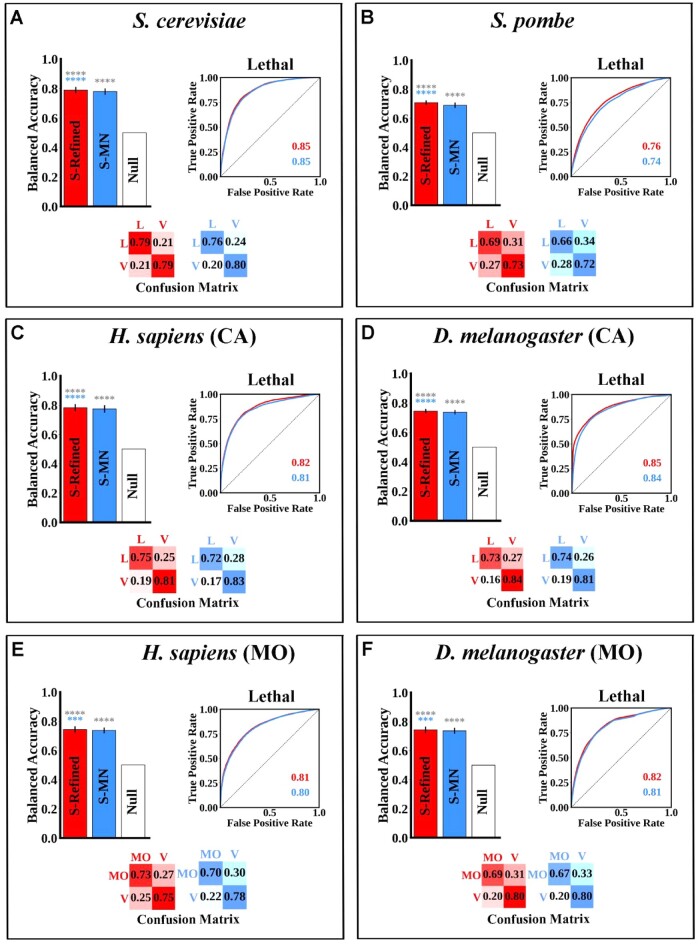
Existing computational models can predict single- and double-mutant fitness but they do have limitations. First, they are often tested via evaluation metrics that are inappropriate for imbalanced datasets. Second, all of them only predict a binary outcome (viable or not, and negatively interacting or not). Third, most are uninterpretable black box machine learning models.
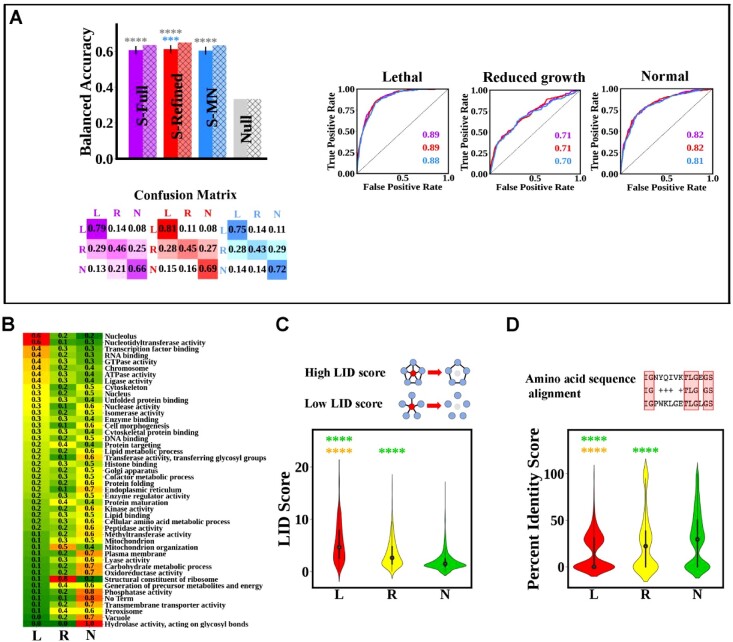
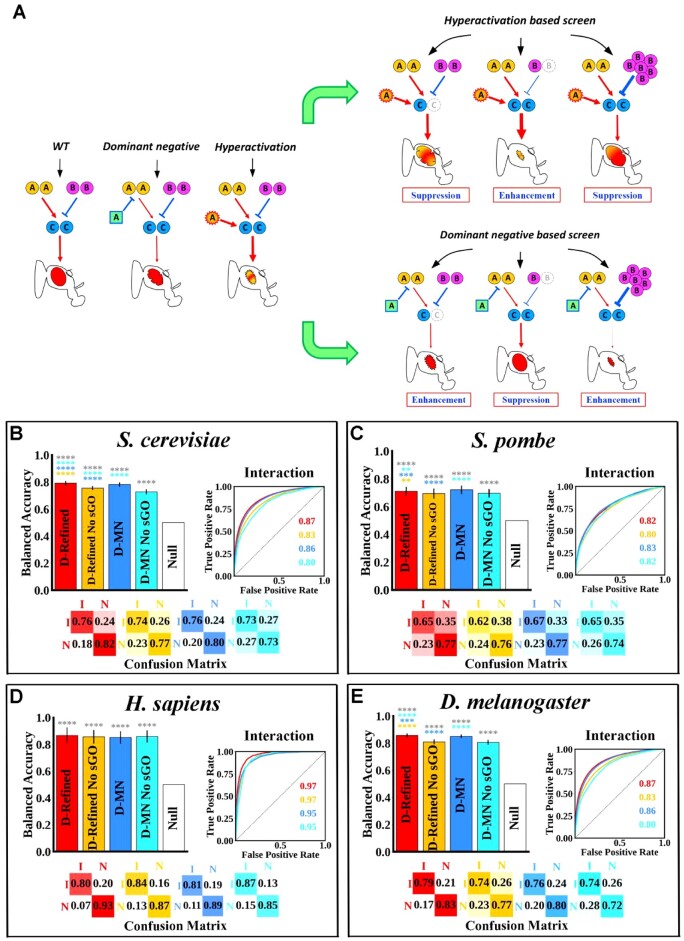
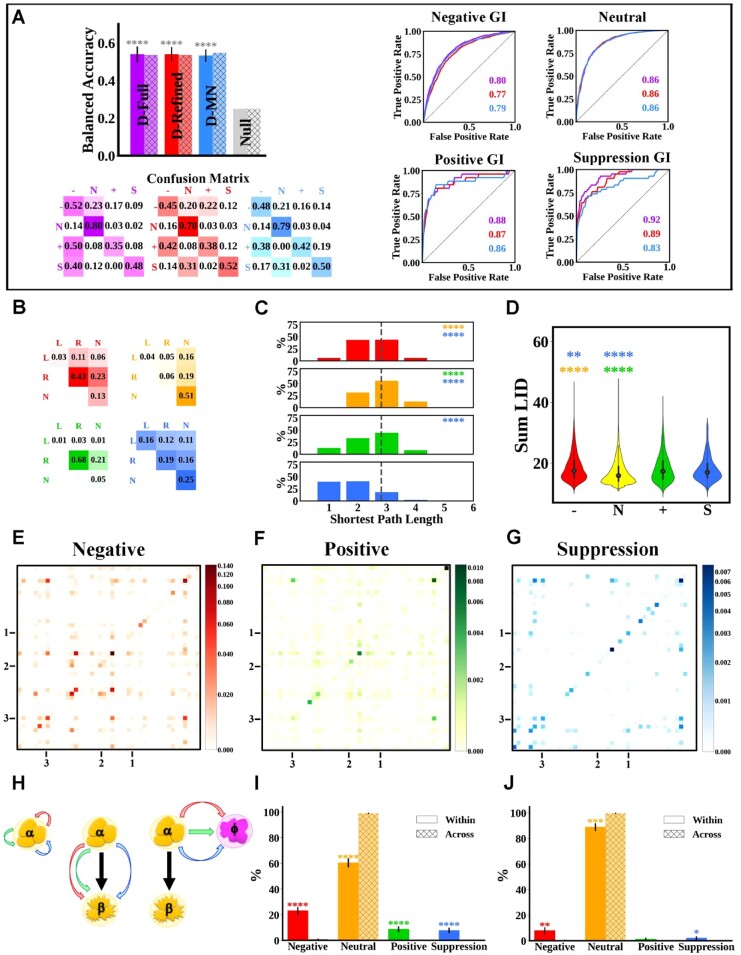
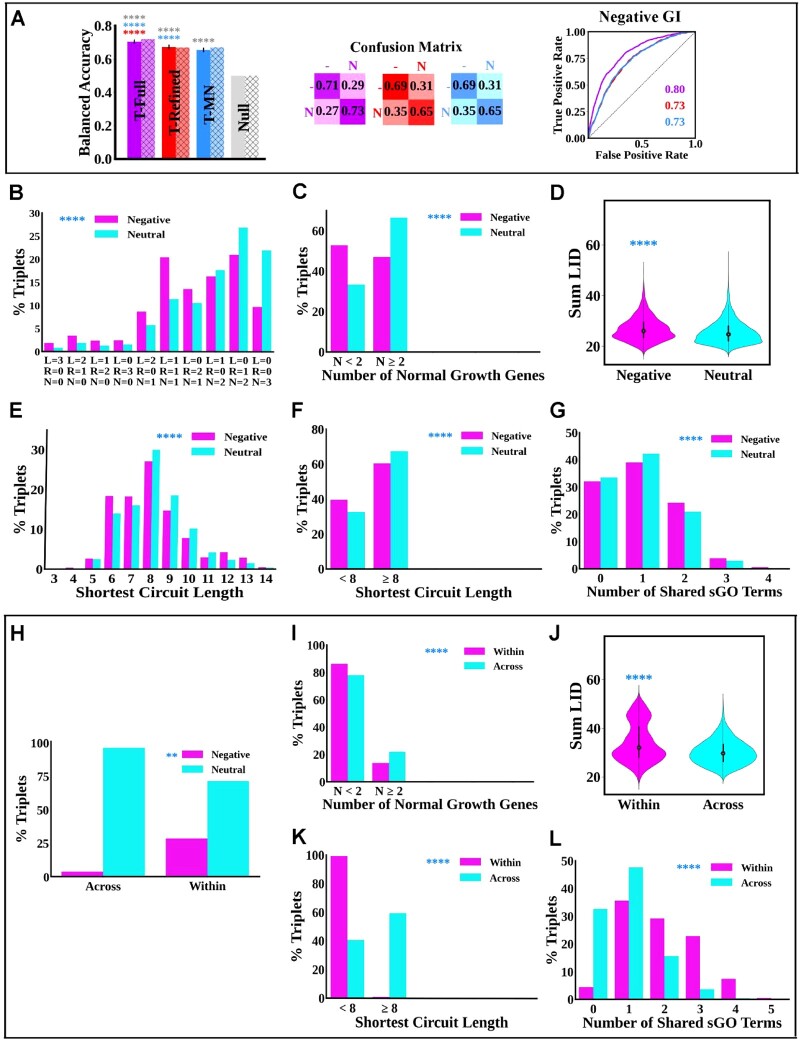
Budding yeast datasets were used to develop high-performance Multinomial Regression (MN) models capable of predicting the impact of single, double and triple genetic disruptions on viability. These models are interpretable and give realistic non-binary predictions and can predict negative genetic interactions (GIs) in triple-gene knockouts. They are based on a limited set of gene features and their predictions are influenced by the probability of target gene participating in molecular complexes or pathways. Furthermore, the MN models have utility in other organisms such as fission yeast, fruit flies and humans, with the single gene fitness MN model being able to distinguish essential genes necessary for cell-autonomous viability from those required for multicellular survival. Finally, our models exceed the performance of previous models, without sacrificing interpretability.
All code and processed datasets used to generate results and figures in this manuscript are available at our Github repository at https://github.com/KISRDevelopment/cell_viability_paper. The repository also contains a link to the GI prediction website that lets users search for GIs using the MN models.
Supplementary data are available at Bioinformatics online.
现有的计算模型可以预测单突变体和双突变体的适应度,但它们确实存在一些局限性。首先,它们通常通过不适合不平衡数据集的评估指标进行测试。其次,它们都只能预测二进制结果(可行或不可行,以及是否存在负相互作用)。第三,大多数都是不可解释的黑盒机器学习模型。
我们使用酿酒酵母数据集开发了高性能多项回归(MN)模型,能够预测单、双和三基因突变对生存能力的影响。这些模型是可解释的,给出了现实的非二进制预测,并可以预测三基因敲除中的负遗传相互作用(GI)。它们基于一组有限的基因特征,其预测受到目标基因参与分子复合物或途径的概率的影响。此外,MN 模型在其他生物体中也具有实用性,例如裂殖酵母、果蝇和人类,单基因适应度 MN 模型能够区分对细胞自主生存至关重要的基因与对多细胞生存所必需的基因。最后,我们的模型在不牺牲可解释性的情况下,超过了以前模型的性能。
本研究手稿中生成结果和图的所有代码和处理数据集都可在我们的 Github 存储库中获得,网址为 https://github.com/KISRDevelopment/cell_viability_paper。该存储库还包含一个 GI 预测网站的链接,用户可以使用 MN 模型搜索 GI。
补充数据可在生物信息学在线获得。