Ni Cheng, Chen Yongjian, Xu Yinchuan, Zhao Jing, Li Qingju, Xiao Changchen, Wu Yan, Wang Jingyi, Wang Yingchao, Zhong Zhiwei, Zhang Ling, Wu Rongrong, Liu Qingnian, Wu Xianpeng, Ke Changle, Zhu Wei, Chen Jinghai, Huang Jijun, Wang Yibin, Wang Jian'an, Hu Xinyang
Department of Cardiology, Cardiovascular Key Laboratory of Zhejiang Province, The Second Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, People's Republic of China (C.N., Y.C., Y.X., J.Z., Q. Li, C.X., Y. Wu, Jingyi Wang, Yingchao Wang, Z.Z., L.Z., R.W., Q. Liu, X.W., C.K., W.Z., J.C., Jian'an Wang, X.H.).
Division of Molecular Medicine, Department of Anesthesiology, David Geffen School of Medicine, University of California at Los Angeles (J.H.).
Circ Res. 2022 Jul 5:101161CIRCRESAHA122320538. doi: 10.1161/CIRCRESAHA.122.320538.
Cardiac fibrosis is a common pathological feature associated with adverse clinical outcome in postinjury remodeling and has no effective therapy. Using an unbiased transcriptome analysis, we identified FMO2 (flavin-containing monooxygenase 2) as a top-ranked gene dynamically expressed following myocardial infarction (MI) in hearts across different species including rodents, nonhuman primates, and human. However, the functional role of FMO2 in cardiac remodeling is largely unknown.
Single-nuclei transcriptome analysis was performed to identify FMO2 after MI; FMO2 ablation rats were generated both in genetic level using the CRISPR-cas9 (clustered regularly interspaced short palindromic repeats/clustered regularly interspaced short palindromic repeat-associated 9) technology and lentivirus-mediated manner. Gain-of-function experiments were conducted using -promoter FMO2, miR1a/miR133a-FMO2 lentivirus, and enzymatic activity mutant FMO2 lentivirus after MI.
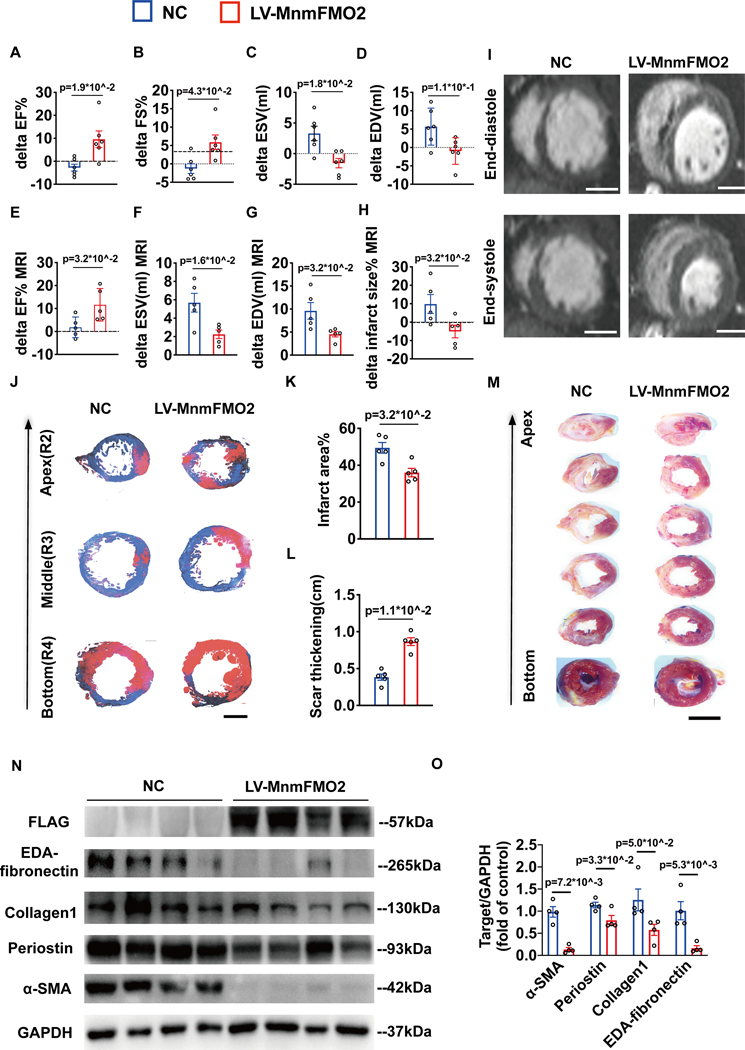
A significant downregulation of FMO2 was consistently observed in hearts after MI in rodents, nonhuman primates, and patients. Single-nuclei transcriptome analysis showed cardiac expression of FMO2 was enriched in fibroblasts rather than myocytes. Elevated spontaneous tissue fibrosis was observed in the FMO2-null animals without external stress. In contrast, fibroblast-specific expression of FMO2 markedly reduced cardiac fibrosis following MI in rodents and nonhuman primates associated with diminished SMAD2/3 phosphorylation. Unexpectedly, the FMO2-mediated regulation in fibrosis and SMAD2/3 signaling was independent of its enzymatic activity. Rather, FMO2 was detected to interact with CYP2J3 (cytochrome p450 superfamily 2J3). Binding of FMO2 to CYP2J3 disrupted CYP2J3 interaction with SMURF2 (SMAD-specific E3 ubiquitin ligase 2) in cytosol, leading to increased cytoplasm to nuclear translocation of SMURF2 and consequent inhibition of SMAD2/3 signaling.
Loss of FMO2 is a conserved molecular signature in postinjury hearts. FMO2 possesses a previously uncharacterized enzyme-independent antifibrosis activity via the CYP2J3-SMURF2 axis. Restoring FMO2 expression exerts potent ameliorative effect against fibrotic remodeling in postinjury hearts from rodents to nonhuman primates. Therefore, FMO2 is a potential therapeutic target for treating cardiac fibrosis following injury.
心脏纤维化是损伤后重塑过程中与不良临床结局相关的常见病理特征,且尚无有效治疗方法。通过无偏转录组分析,我们确定含黄素单加氧酶2(FMO2)是在包括啮齿动物、非人灵长类动物和人类在内的不同物种心脏发生心肌梗死后动态表达的排名靠前的基因。然而,FMO2在心脏重塑中的功能作用在很大程度上尚不清楚。
进行单核转录组分析以确定心肌梗死后的FMO2;使用CRISPR - cas9(规律成簇间隔短回文重复序列/规律成簇间隔短回文重复序列相关蛋白9)技术和慢病毒介导的方式在基因水平上构建FMO2基因敲除大鼠。心肌梗死后,使用FMO2启动子、miR1a/miR133a - FMO2慢病毒和酶活性突变体FMO2慢病毒进行功能获得实验。
在啮齿动物、非人灵长类动物和患者心肌梗死后的心脏中,一致观察到FMO2显著下调。单核转录组分析表明,FMO2在心脏中的表达在成纤维细胞中富集,而非心肌细胞。在无外部应激的FMO2基因敲除动物中观察到自发性组织纤维化增加。相反,在啮齿动物和非人灵长类动物中,心肌梗死后FMO2在成纤维细胞中的特异性表达显著减少了心脏纤维化,这与SMAD2/3磷酸化减少有关。出乎意料的是,FMO2在纤维化和SMAD2/3信号传导中的调节作用与其酶活性无关。相反,检测到FMO2与细胞色素P450超家族2J3(CYP2J3)相互作用。FMO2与CYP2J3的结合破坏了CYP2J3与胞质溶胶中SMAD特异性E3泛素连接酶2(SMURF2)的相互作用,导致SMURF2从细胞质向细胞核的转位增加,从而抑制SMAD2/3信号传导。
FMO2缺失是损伤后心脏中保守的分子特征。FMO2通过CYP2J3 - SMURF2轴具有一种以前未被描述的非酶依赖性抗纤维化活性。恢复FMO2表达对从啮齿动物到非人灵长类动物损伤后心脏的纤维化重塑具有强大的改善作用。因此,FMO2是治疗损伤后心脏纤维化的潜在治疗靶点。