Inserm, BRM (Bacterial Regulatory RNAs and Medicine)-UMR_S 1230, Rennes, France.
Institut Agro, CNRS, Université de Rennes, IRMAR (Institut de Recherche Mathématique de Rennes)-UMR 6625, Rennes, France.
mSystems. 2022 Aug 30;7(4):e0037822. doi: 10.1128/msystems.00378-22. Epub 2022 Jul 5.
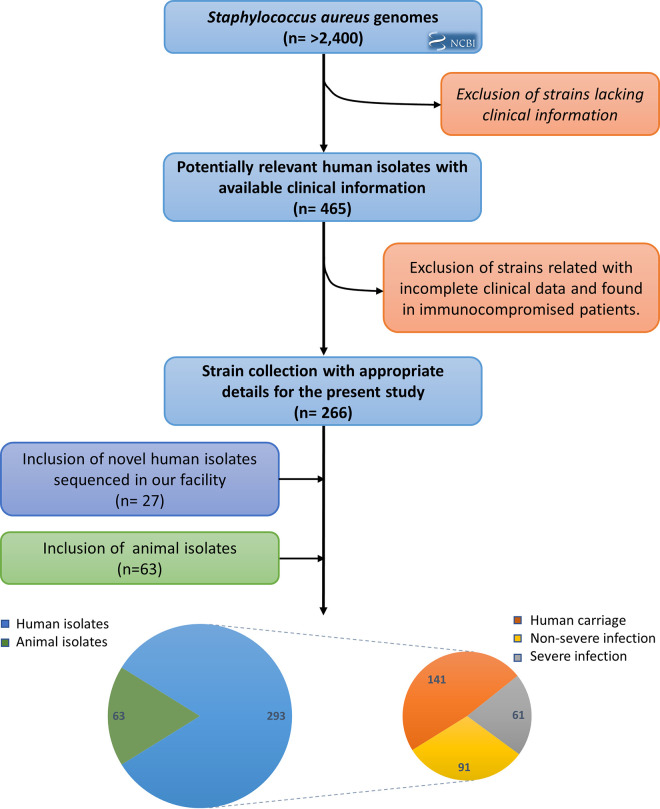
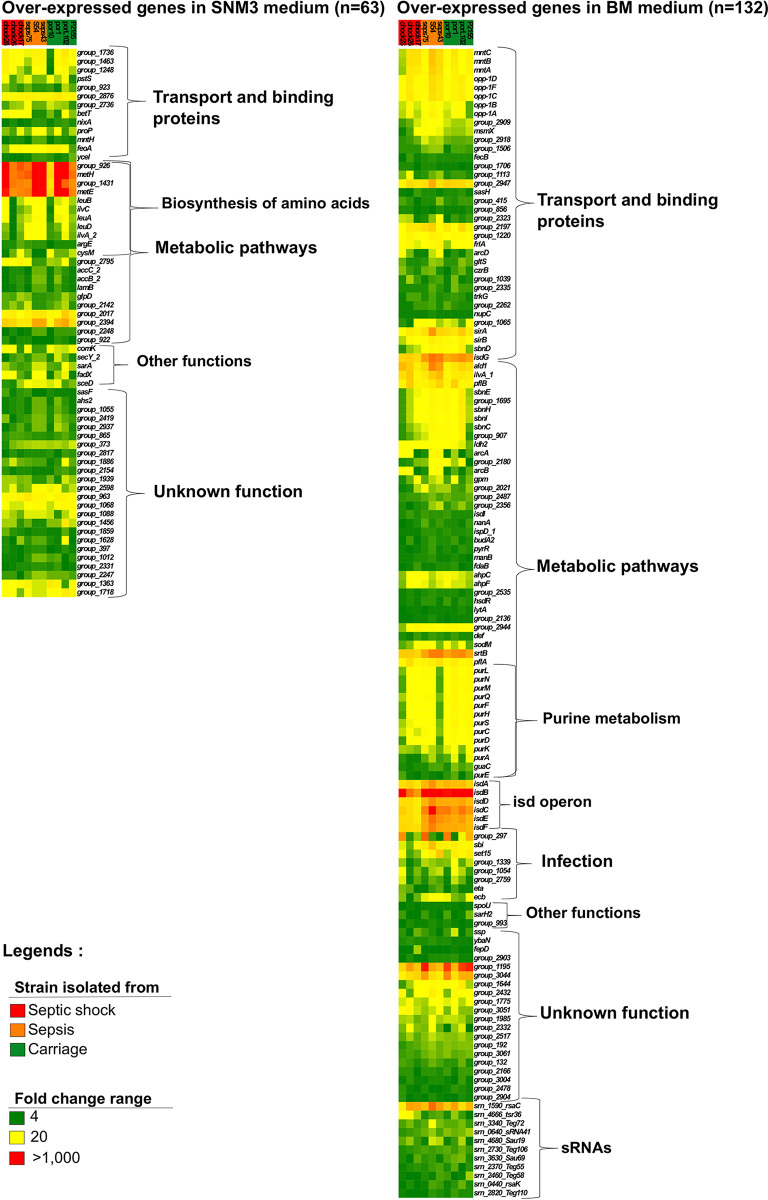
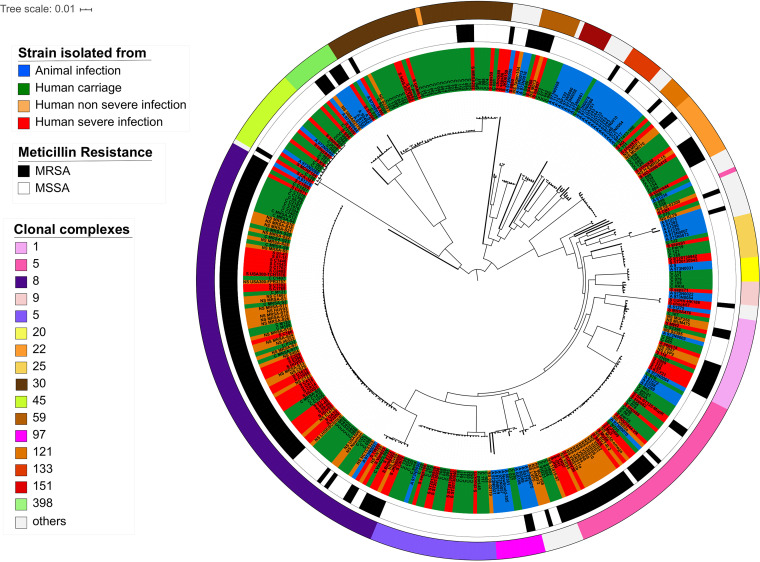
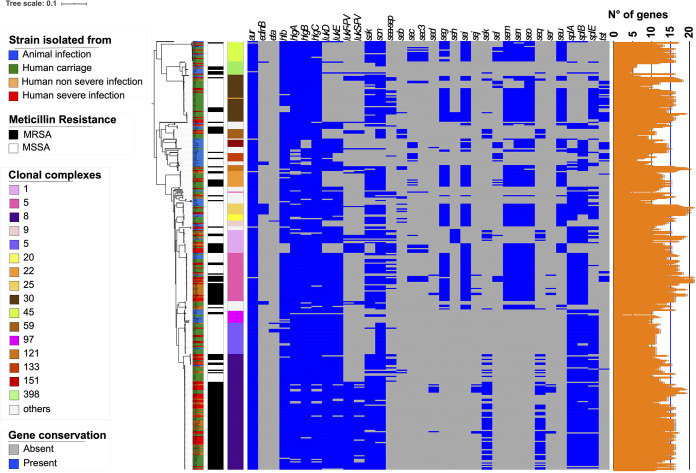
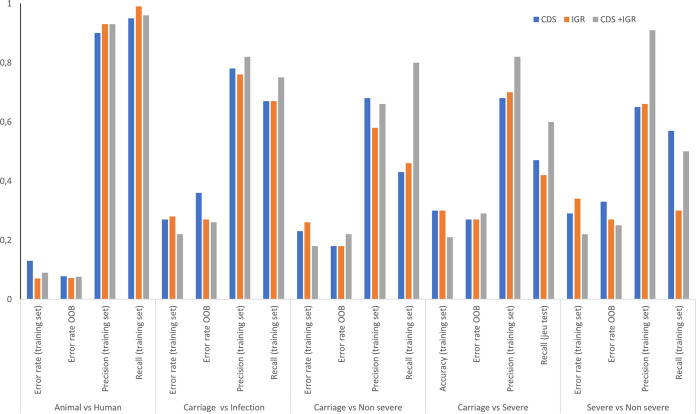
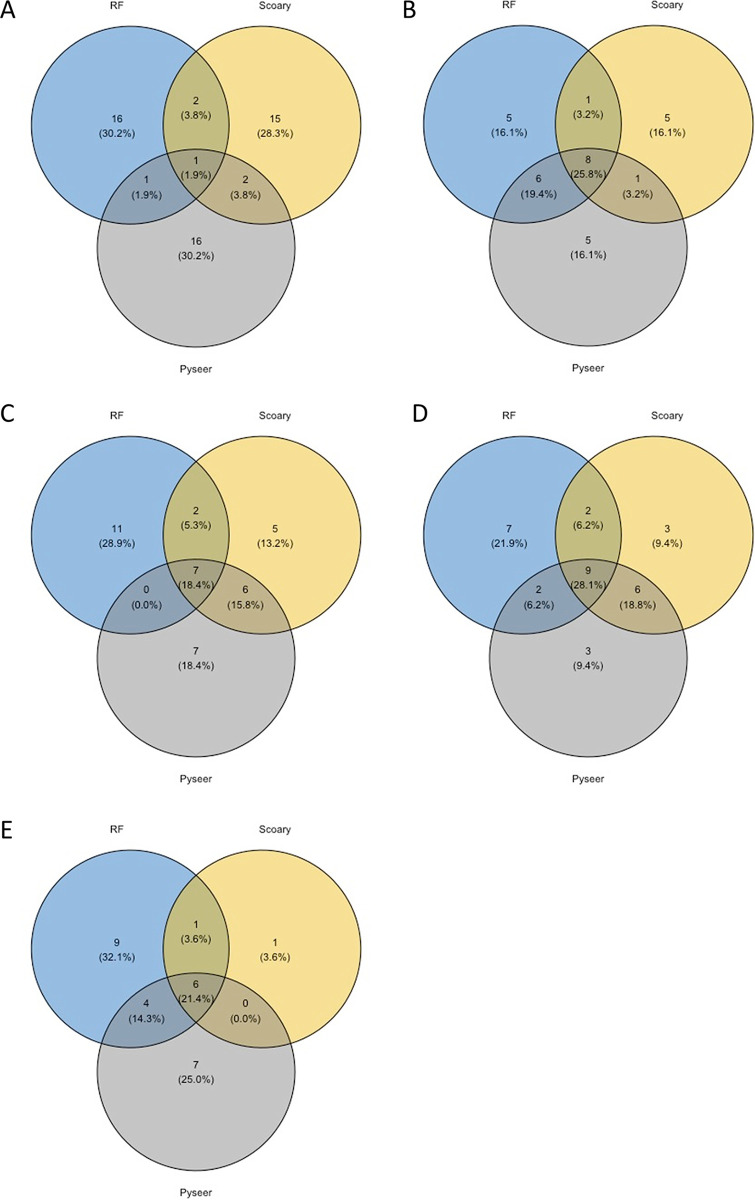
Staphylococcus aureus is a major human and animal pathogen, colonizing diverse ecological niches within its hosts. Predicting whether an isolate will infect a specific host and its subsequent clinical fate remains unknown. In this study, we investigated the S. aureus pangenome using a curated set of 356 strains, spanning a wide range of hosts, origins, and clinical display and antibiotic resistance profiles. We used genome-wide association study (GWAS) and random forest (RF) algorithms to discriminate strains based on their origins and clinical sources. Here, we show that the presence of and can discriminate strains based on their host specificity, while other genes such as are often associated with virulent outcomes. Both GWAS and RF indicated the importance of intergenic regions (IGRs) and coding DNA sequence (CDS) but not sRNAs in forecasting an outcome. Additional transcriptomic analyses performed on the most prevalent clonal complex 8 (CC8) clonal types, in media mimicking nasal colonization or bacteremia, indicated three RNAs as potential RNA markers to forecast infection, followed by 30 others that could serve as infection severity predictors. Our report shows that genetic association and transcriptomics are complementary approaches that will be combined in a single analytical framework to improve our understanding of bacterial pathogenesis and ultimately identify potential predictive molecular markers. Predicting the outcome of bacterial colonization and infections, based on extensive genomic and transcriptomic data from a given pathogen, would be of substantial help for clinicians in treating and curing patients. In this report, genome-wide association studies and random forest algorithms have defined gene combinations that differentiate human from animal strains, colonization from diseases, and nonsevere from severe diseases, while it revealed the importance of IGRs and CDS, but not small RNAs (sRNAs), in anticipating an outcome. In addition, transcriptomic analyses performed on the most prevalent clonal types, in media mimicking either nasal colonization or bacteremia, revealed significant differences and therefore potent RNA markers. Overall, the use of both genomic and transcriptomic data in a single analytical framework can enhance our understanding of bacterial pathogenesis.
金黄色葡萄球菌是一种主要的人类和动物病原体,定植于宿主内的多种生态位。预测分离株是否会感染特定宿主及其随后的临床转归仍然未知。在这项研究中,我们使用经过精心挑选的 356 株菌株研究了金黄色葡萄球菌的泛基因组,这些菌株涵盖了广泛的宿主、来源以及临床表型和抗生素耐药谱。我们使用全基因组关联研究(GWAS)和随机森林(RF)算法根据菌株的来源和临床来源对其进行区分。在这里,我们表明 和 的存在可以根据宿主特异性来区分菌株,而其他基因,如 ,通常与毒力结果相关。GWAS 和 RF 都表明,种间区(IGR)和编码 DNA 序列(CDS)的重要性,但非小 RNA(sRNA)在预测结果中并不重要。在最常见的克隆复合体 8(CC8)克隆型的模拟鼻腔定植或菌血症的培养基上进行的额外转录组分析表明,三种 RNA 可能作为预测感染的 RNA 标志物,随后有 30 种 RNA 可能作为感染严重程度的预测因子。我们的报告表明,遗传关联和转录组学是互补的方法,将在单个分析框架中结合使用,以提高我们对细菌发病机制的理解,并最终确定潜在的预测分子标志物。基于给定病原体的广泛基因组和转录组数据预测细菌定植和感染的结果,将对临床医生治疗和治愈患者有很大帮助。在本报告中,全基因组关联研究和随机森林算法定义了区分人源和动物源菌株、定植和疾病以及非严重和严重疾病的基因组合,同时揭示了 IGR 和 CDS 的重要性,而不是小 RNA(sRNA)在预测结果中的重要性。此外,在模拟鼻腔定植或菌血症的培养基上对最常见的克隆型进行的转录组分析揭示了显著差异,因此存在有效的 RNA 标志物。总的来说,在单个分析框架中同时使用基因组和转录组数据可以增强我们对细菌发病机制的理解。