Wirth Thierry, Wong Vanessa, Vandenesch François, Rasigade Jean-Philippe
Institut de Systématique, Evolution, Biodiversité UMR-CNRS 7205 Muséum National d'Histoire Naturelle Université Pierre et Marie Curie Université des Antilles Ecole Pratique des Hautes Etudes Sorbonne Universités Paris France.
EPHE PSL University Paris France.
Evol Appl. 2020 May 22;13(6):1513-1525. doi: 10.1111/eva.12991. eCollection 2020 Jul.
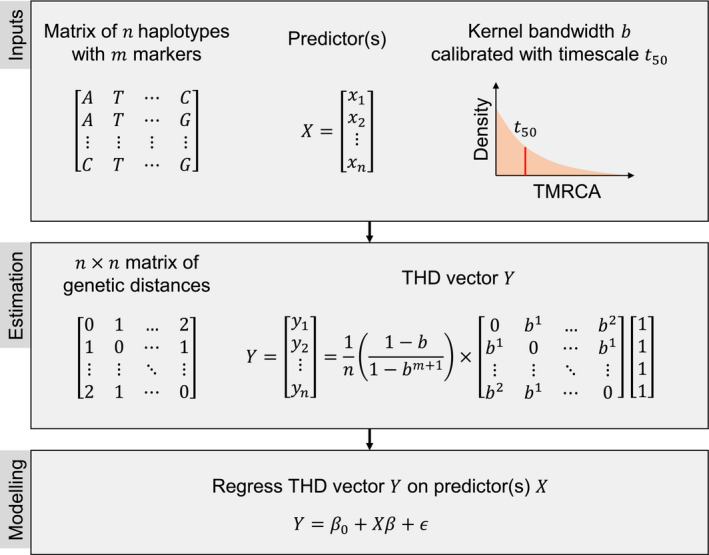
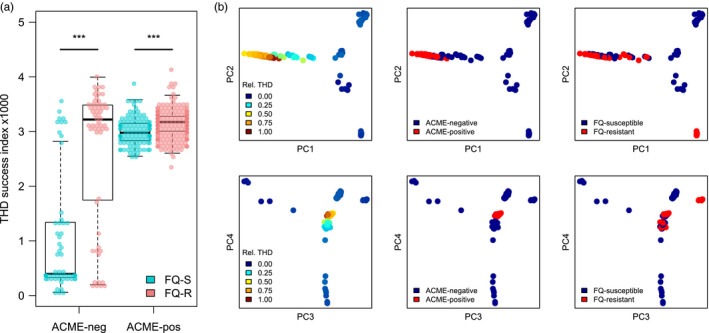
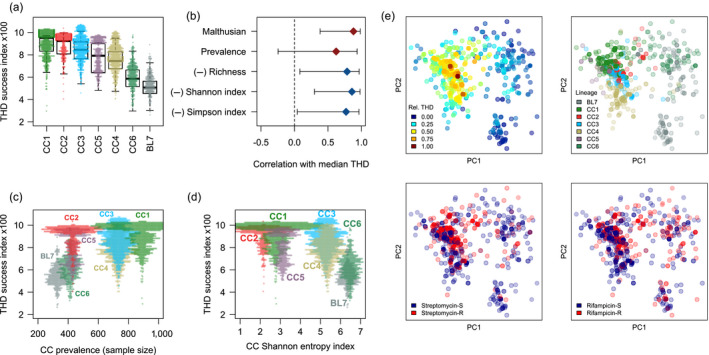
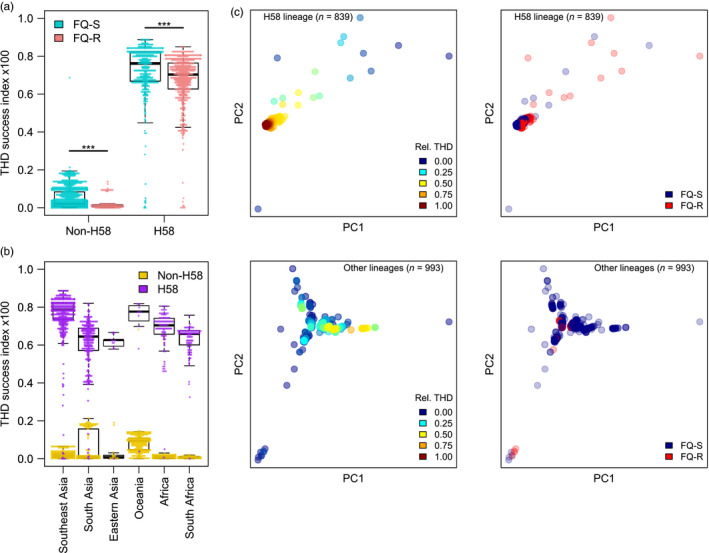
Understanding the driving forces of an epidemic is key to inform intervention strategies against it. Correlating measures of the epidemic success of a pathogen with ancillary parameters such as its drug resistance profile provides a flexible tool to identify such driving forces. The recently described time-scaled haplotypic density (THD) method facilitates the inference of a pathogen's epidemic success from genetic data. Contrary to demogenetic approaches that define success in an aggregated fashion, the THD computes an independent index of success for each isolate in a collection. Modeling this index using multivariate regression, thus, allows us to control for various sources of bias and to identify independent predictors of success. We illustrate the use of THD to address key questions regarding three exemplary epidemics of multidrug-resistant (MDR) bacterial lineages, namely Beijing, Typhi H58, and ST8 (including ST8-USA300 MRSA), based on previously published, international genetic datasets. In each case, THD analysis allowed to identify the impact, or lack thereof, of various factors on the epidemic success, independent of confounding by population structure and geographic distribution. Our results suggest that rifampicin resistance drives the MDR Beijing epidemic and that fluoroquinolone resistance drives the ST8/USA300 epidemic, in line with previous evidence of a lack of resistance-associated fitness cost in these pathogens. Conversely, fluoroquinolone resistance measurably hampered the success of Typhi H58 and non-H58. These findings illustrate how THD can help leverage the massive genomic datasets generated by molecular epidemiology studies to address new questions. THD implementation for the R platform is available at https://github.com/rasigadelab/thd.
了解流行病的驱动因素是制定防控策略的关键。将病原体在流行过程中的传播情况与诸如耐药谱等辅助参数相关联,可为识别这些驱动因素提供一种灵活的工具。最近描述的时间尺度单倍型密度(THD)方法有助于从基因数据推断病原体在流行中的传播情况。与以聚合方式定义传播成功的群体遗传学方法不同,THD为数据集中的每个分离株计算一个独立的传播成功指数。因此,使用多元回归对该指数进行建模,使我们能够控制各种偏差来源,并识别传播成功的独立预测因素。我们基于先前发表的国际基因数据集,举例说明了如何使用THD来解决关于三种耐多药(MDR)细菌谱系(即北京型、伤寒H58型和ST8型,包括ST8-USA300耐甲氧西林金黄色葡萄球菌)典型流行的关键问题。在每种情况下,THD分析都能够识别各种因素对流行传播成功的影响或无影响,而不受群体结构和地理分布的混杂影响。我们的结果表明,利福平耐药推动了耐多药北京型菌株的流行,氟喹诺酮耐药推动了ST8/USA300菌株的流行,这与之前关于这些病原体中缺乏耐药相关适应性代价的证据一致。相反,氟喹诺酮耐药显著阻碍了伤寒H58型和非H58型菌株的传播成功。这些发现说明了THD如何有助于利用分子流行病学研究产生的大量基因组数据集来解决新问题。R平台的THD实现可在https://github.com/rasigadelab/thd获取。