Pavan Matteo, Menin Silvia, Bassani Davide, Sturlese Mattia, Moro Stefano
Molecular Modeling Section (MMS), Department of Pharmaceutical and Pharmacological Sciences, University of Padova, Padova, Italy.
Front Mol Biosci. 2022 Jul 7;9:909499. doi: 10.3389/fmolb.2022.909499. eCollection 2022.
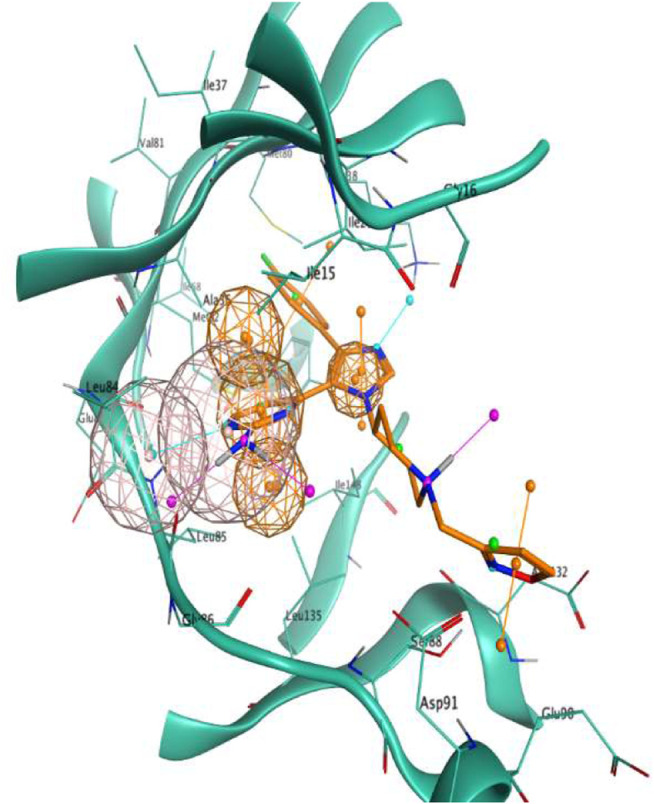
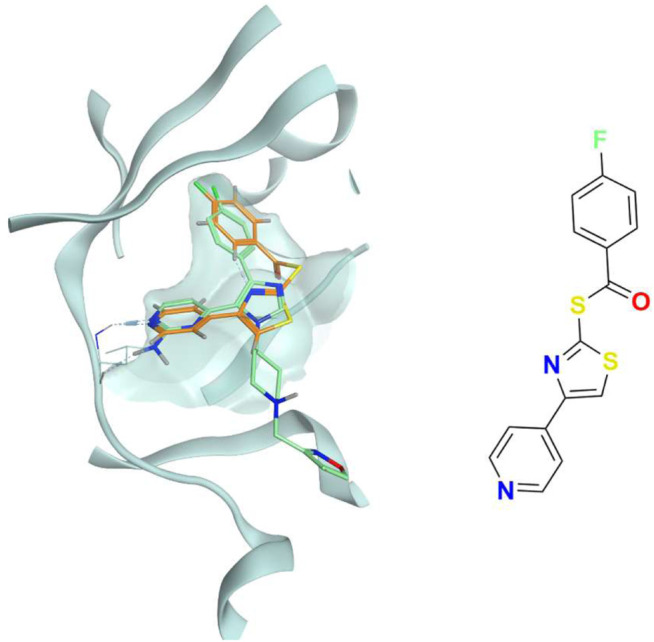
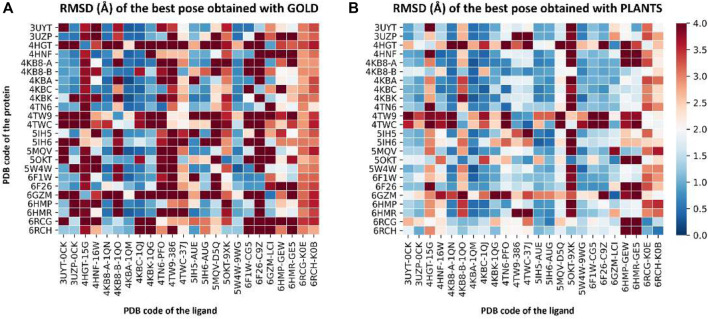
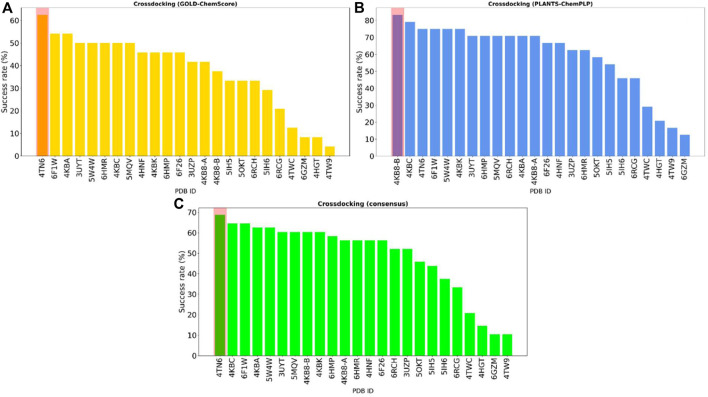
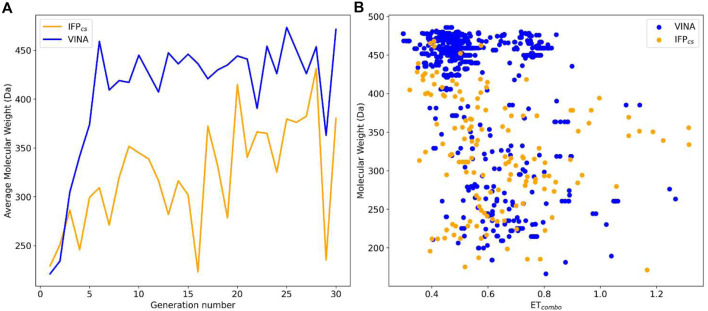
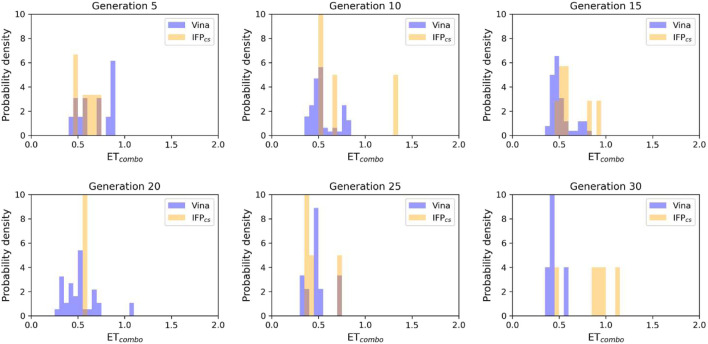
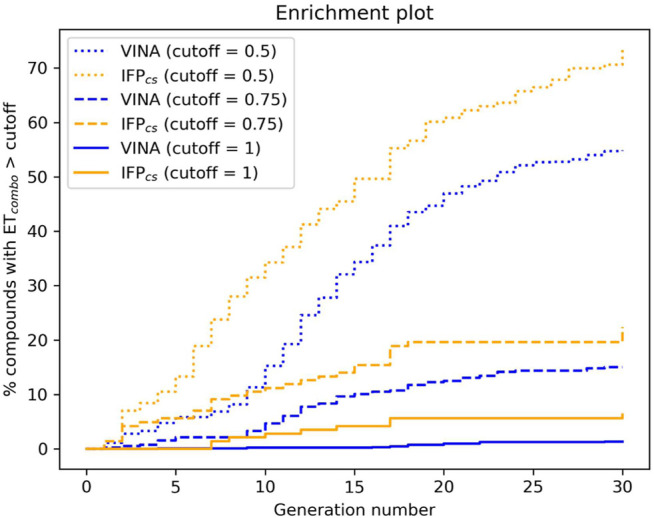
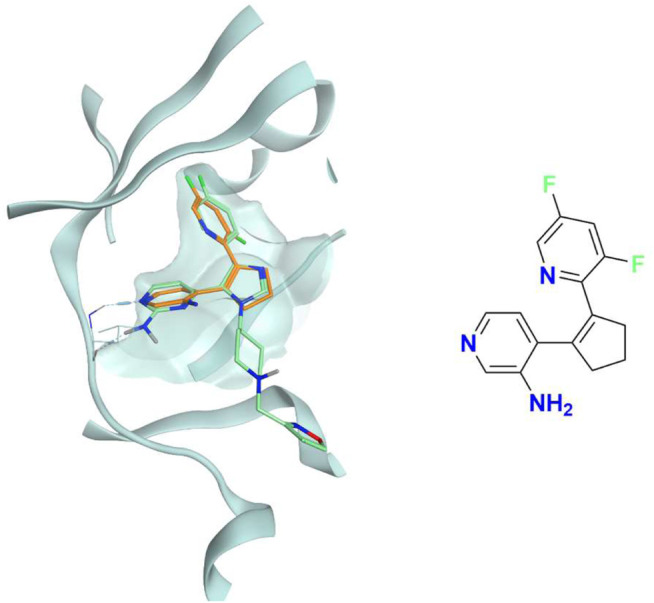
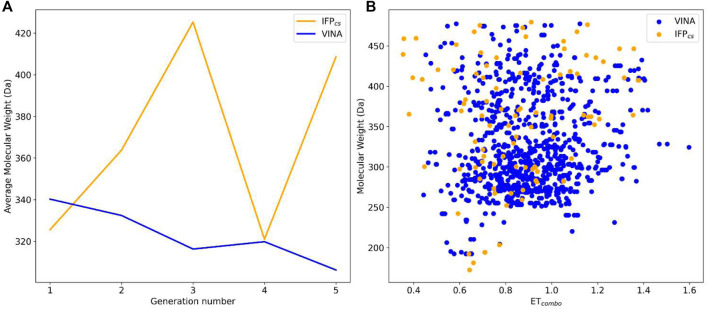
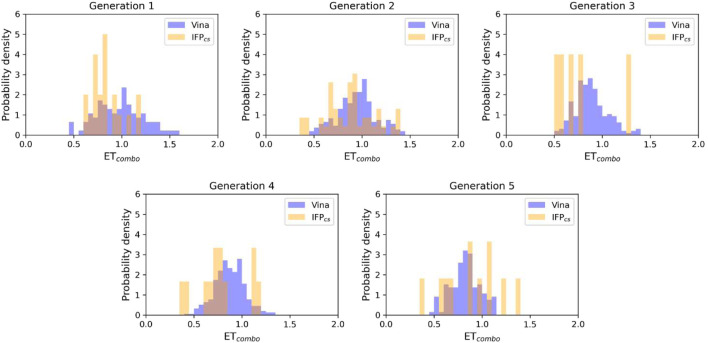
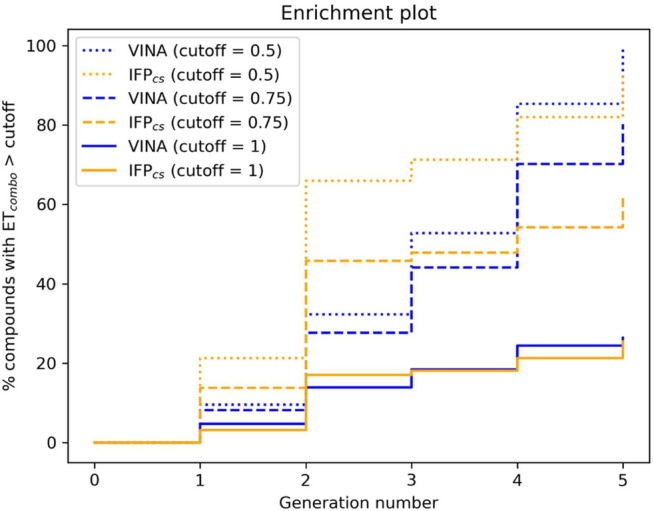
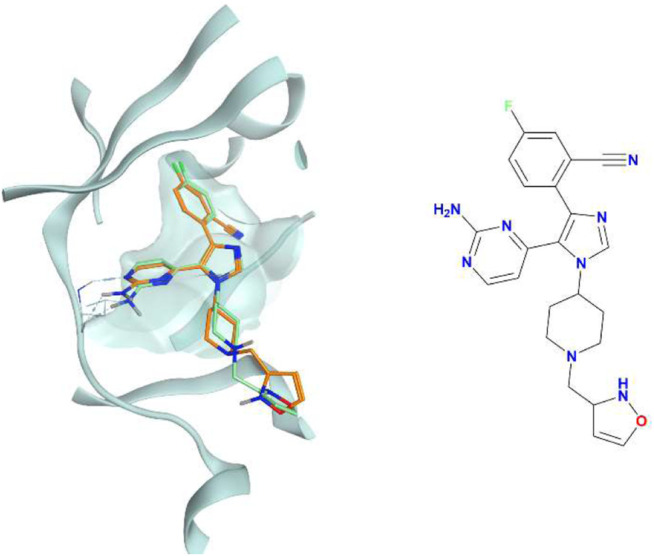
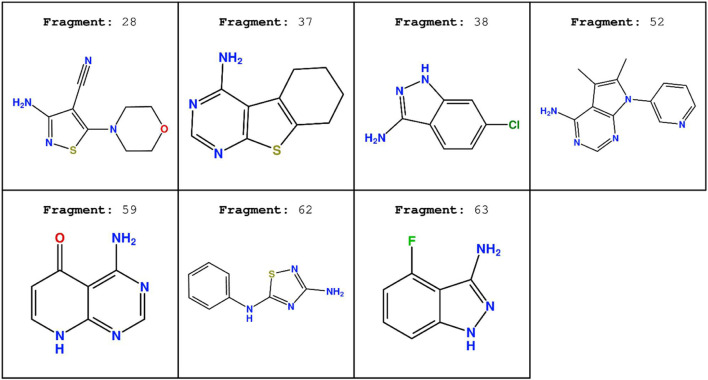
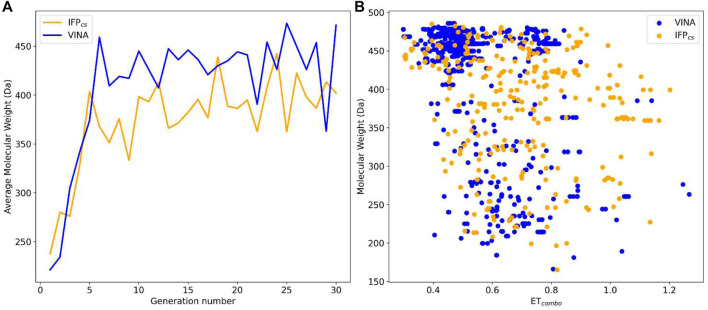
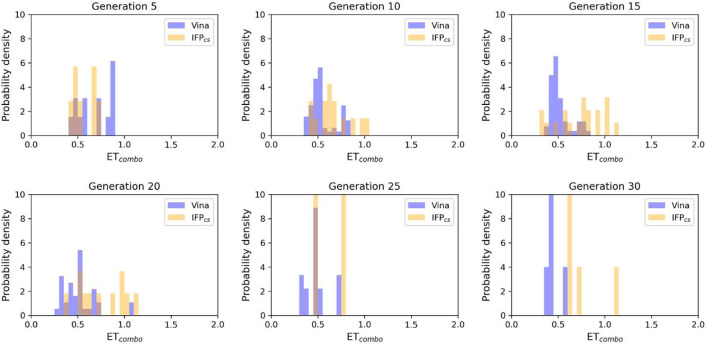
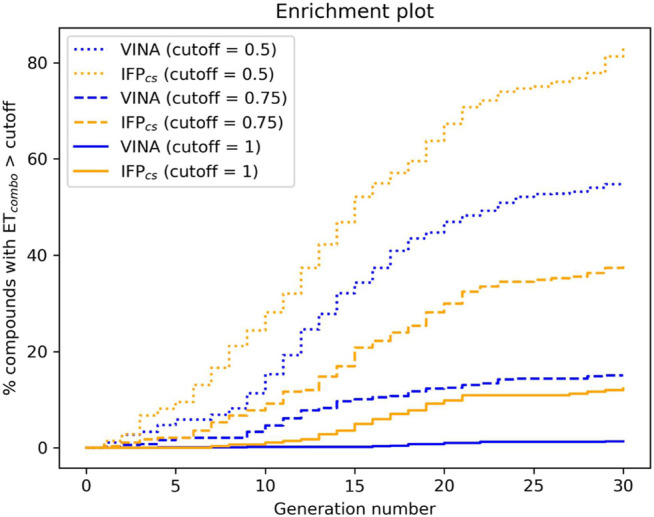
In the last 20 years, fragment-based drug discovery (FBDD) has become a popular and consolidated approach within the drug discovery pipeline, due to its ability to bring several drug candidates to clinical trials, some of them even being approved and introduced to the market. A class of targets that have proven to be particularly suitable for this method is represented by kinases, as demonstrated by the approval of BRAF inhibitor vemurafenib. Within this wide and diverse set of proteins, protein kinase CK1δ is a particularly interesting target for the treatment of several widespread neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis. Computational methodologies, such as molecular docking, are already routinely and successfully applied in FBDD campaigns alongside experimental techniques, both in the hit-discovery and in the hit-optimization stage. Concerning this, the open-source software Autogrow, developed by the Durrant lab, is a semi-automated computational protocol that exploits a combination between a genetic algorithm and a molecular docking software for drug design and lead optimization. In the current work, we present and discuss a modified version of the Autogrow code that implements a custom scoring function based on the similarity between the interaction fingerprint of investigated compounds and a crystal reference. To validate its performance, we performed both a and a lead-optimization run (as described in the original publication), evaluating the ability of our fingerprint-based protocol to generate compounds similar to known CK1δ inhibitors based on both the predicted binding mode and the electrostatic and shape similarity in comparison with the standard Autogrow protocol.
在过去20年中,基于片段的药物发现(FBDD)已成为药物发现流程中一种流行且成熟的方法,因为它有能力将多个候选药物推进到临床试验阶段,其中一些甚至已获批并推向市场。激酶类靶点已被证明特别适合这种方法,BRAF抑制剂维莫非尼的获批就证明了这一点。在这一广泛且多样的蛋白质组中,蛋白激酶CK1δ是治疗多种常见神经退行性疾病(如阿尔茨海默病、帕金森病和肌萎缩侧索硬化症)的一个特别有趣的靶点。计算方法,如分子对接,已在FBDD研究中与实验技术一起常规且成功地应用于命中发现和命中优化阶段。关于这一点,由杜兰特实验室开发的开源软件Autogrow是一种半自动化计算协议,它利用遗传算法和分子对接软件的组合进行药物设计和先导优化。在当前工作中,我们展示并讨论了Autogrow代码的一个修改版本,该版本基于所研究化合物的相互作用指纹与晶体参考之间的相似性实现了一个自定义评分函数。为了验证其性能,我们进行了一次命中发现运行和一次先导优化运行(如原始出版物中所述),评估了我们基于指纹的协议基于预测的结合模式以及与标准Autogrow协议相比的静电和形状相似性生成与已知CK1δ抑制剂相似的化合物的能力。