Department of Obstetrics and Gynaecology, Shengjing Hospital of China Medical University, Shenyang, China.
Front Immunol. 2022 Jul 8;13:883404. doi: 10.3389/fimmu.2022.883404. eCollection 2022.
Preeclampsia is a common and serious complication of pregnancy, posing a threat to maternal and fetal safety due to the lack of effective biomarkers and treatment strategies. This study aimed to identify potential biomarkers that can be used to predict preeclampsia and identify the molecular mechanisms of preeclampsia pathogenesis and drug prediction at the transcriptome level.
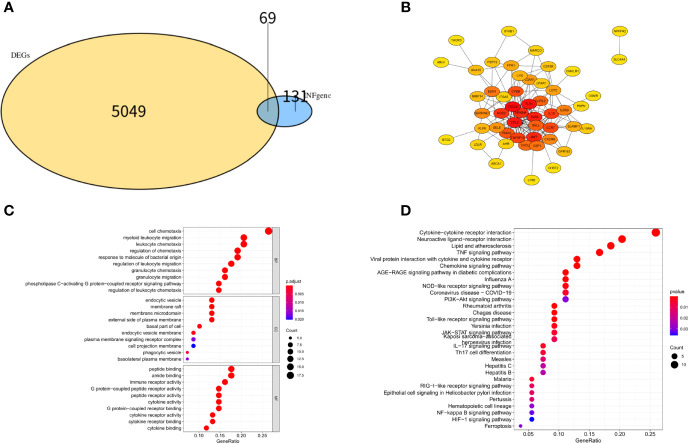
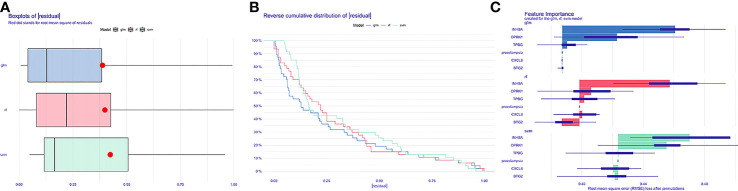
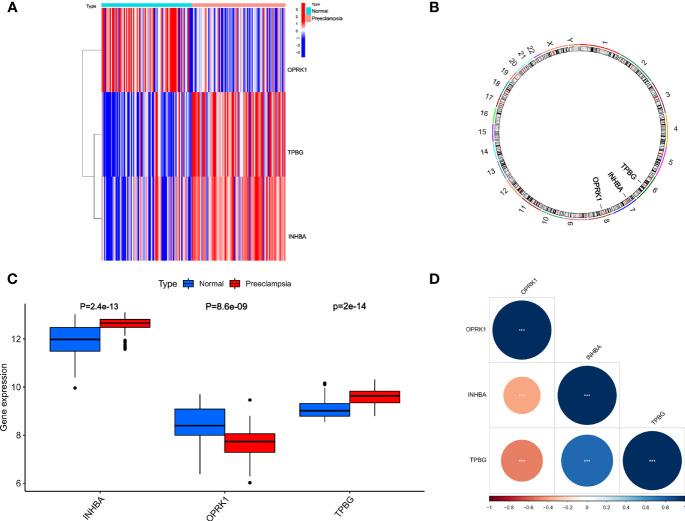
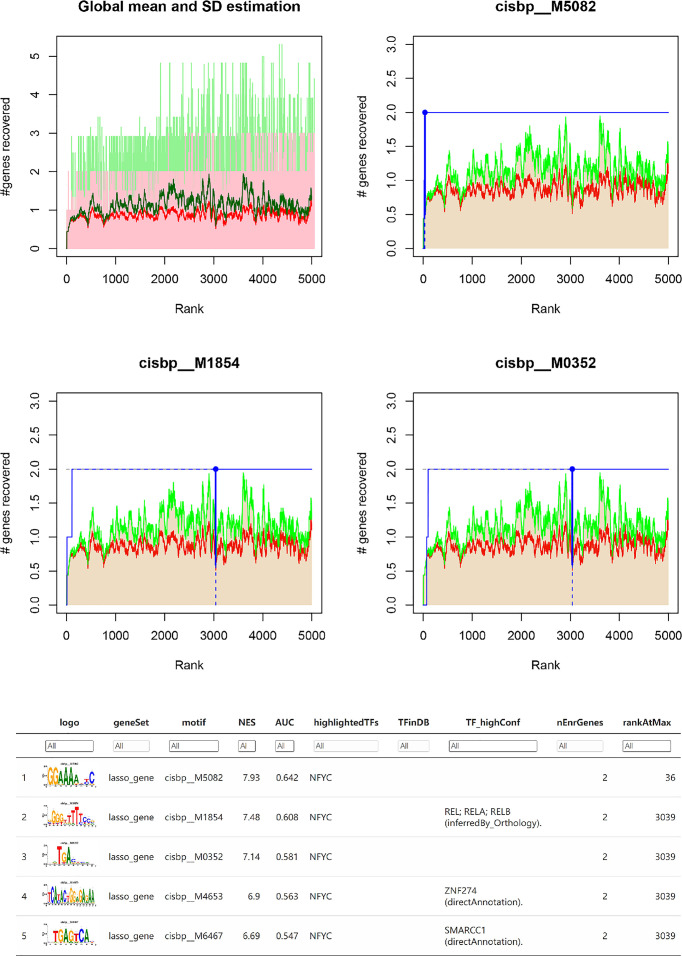
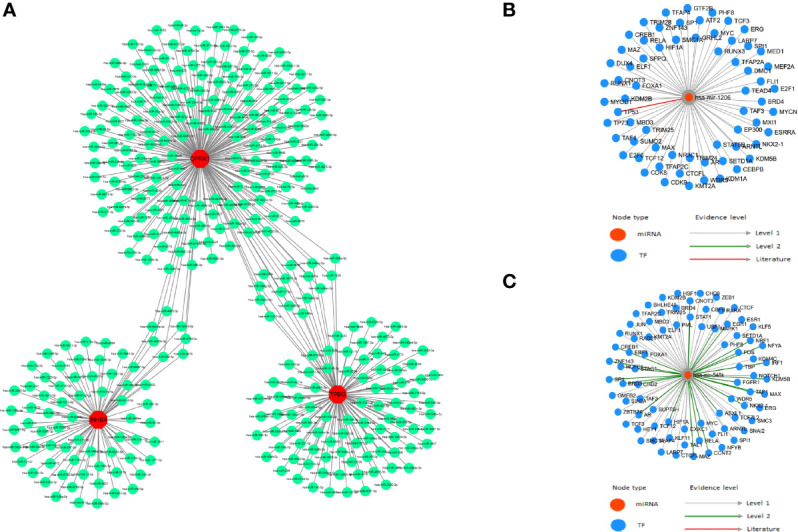
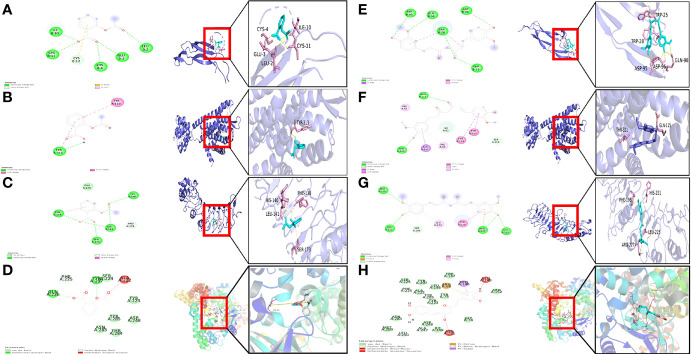
We analyzed differential expression genes (DEGs) in preeclampsia and non-preeclampsia groups in the GSE75010 dataset, cross-linking with extracted inflammatory response-related genes to obtain differentially expressed inflammation-related genes (DINRGs). Enrichment analysis and protein-protein interaction (PPI) networks were constructed to understand the functions and enrichment pathways. Machine learning models were used to identify key genes associated with preeclampsia and build a nomogram in the training set, which was validated in the validation set. The R package RcisTarget was used to predict transcription factors, and Cytoscape was used to construct miRNA-mRNA pathways, which could identify the molecular mechanisms. Then, we conducted molecular docking of the obtained key genes (inhibin subunit beta A), (opioid receptor kappa 1), and (trophoblast glycoprotein), as well as predicted transcription factors with drug molecules. Additionally, the CIBERSORT method explored the differences in immune cell infiltration between preeclampsia and non-preeclampsia samples based on the GSE75010 dataset.
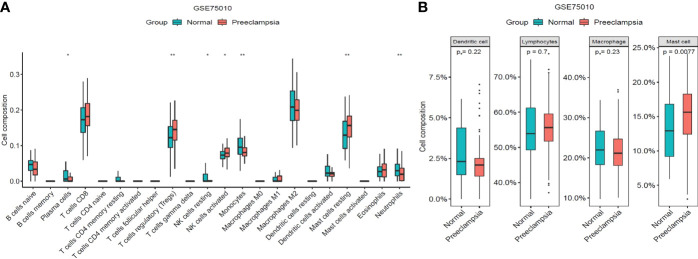
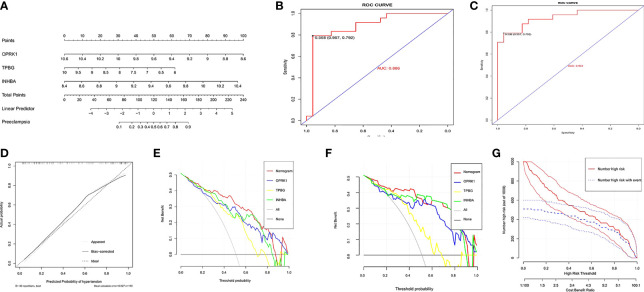
A total of 69 DINRGs associated with preeclampsia patients were screened. , and were the key genes based on machine learning models. A nomogram for prediction was further constructed, and the receiver operating curves (ROCs) showed good performance. Based on the transcriptome level of key genes, we proposed that RELA-miR-548K/miR-1206-TPBG may be a potential RNA regulatory pathway regulating the progression of early preeclampsia. Molecular docking suggested the effectiveness of curcumin in the treatment of preeclampsia. Additionally, regulatory T cells (Tregs) and resting mast cells were significantly different between the two groups.
In summary, we identified three key inflammation-associated genes, namely , and , which can be used as potential genetic biomarkers for preeclampsia prediction and treatment, and established a nomogram as a predictive model. Additionally, we provided insights into the mechanisms of preeclampsia development at the transcriptome level and performed corresponding drug predictions.
子痫前期是一种常见且严重的妊娠并发症,由于缺乏有效的生物标志物和治疗策略,对母婴安全构成威胁。本研究旨在寻找可用于预测子痫前期的潜在生物标志物,并在转录组水平上识别子痫前期发病机制和药物预测的分子机制。
我们分析了 GSE75010 数据集子痫前期和非子痫前期组中的差异表达基因(DEGs),与提取的炎症反应相关基因交联,获得差异表达的炎症反应相关基因(DINRGs)。进行富集分析和蛋白质-蛋白质相互作用(PPI)网络构建,以了解功能和富集途径。使用机器学习模型识别与子痫前期相关的关键基因,并在训练集中构建列线图,在验证集中进行验证。使用 R 包 RcisTarget 预测转录因子,并使用 Cytoscape 构建 miRNA-mRNA 途径,以识别分子机制。然后,我们对获得的关键基因(抑制素亚单位β A)、(阿片受体κ 1)和(滋养细胞糖蛋白)以及预测的转录因子与药物分子进行分子对接。此外,根据 GSE75010 数据集,CIBERSORT 方法探讨了子痫前期和非子痫前期样本中免疫细胞浸润的差异。
筛选出与子痫前期患者相关的共 69 个 DINRGs。基于机器学习模型, 、 和 是关键基因。进一步构建了预测列线图,ROC 表现良好。基于关键基因的转录组水平,我们提出 RELA-miR-548K/miR-1206-TPBG 可能是调节早期子痫前期进展的潜在 RNA 调控途径。分子对接表明姜黄素在子痫前期治疗中的有效性。此外,两组之间调节性 T 细胞(Tregs)和静止肥大细胞存在显著差异。
综上所述,我们鉴定了三个关键的炎症相关基因,即 、 和 ,它们可以作为子痫前期预测和治疗的潜在遗传生物标志物,并建立了预测模型。此外,我们在转录组水平上提供了子痫前期发病机制的见解,并进行了相应的药物预测。