Turku Bioscience Centre, University of Turku and Åbo Akademi University, Tykistökatu 6, 20520 Turku, Finland.
Institute of Biomedicine, University of Turku, 20520 Turku, Finland.
Brief Bioinform. 2022 Sep 20;23(5). doi: 10.1093/bib/bbac286.
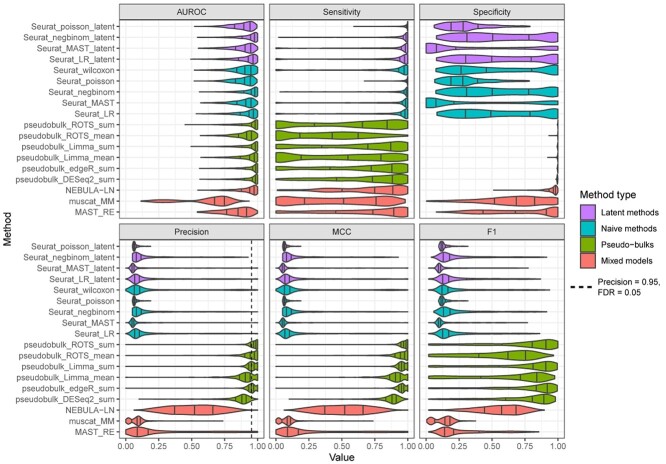
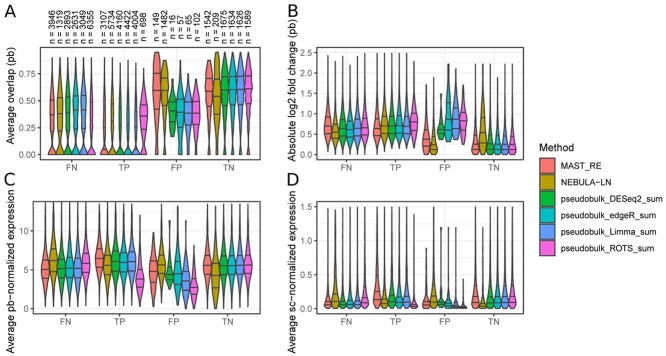
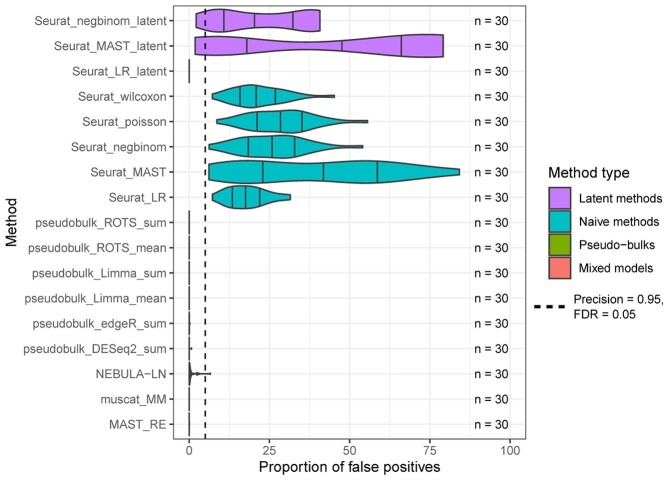
Single-cell RNA-sequencing (scRNA-seq) enables researchers to quantify transcriptomes of thousands of cells simultaneously and study transcriptomic changes between cells. scRNA-seq datasets increasingly include multisubject, multicondition experiments to investigate cell-type-specific differential states (DS) between conditions. This can be performed by first identifying the cell types in all the subjects and then by performing a DS analysis between the conditions within each cell type. Naïve single-cell DS analysis methods that treat cells statistically independent are subject to false positives in the presence of variation between biological replicates, an issue known as the pseudoreplicate bias. While several methods have already been introduced to carry out the statistical testing in multisubject scRNA-seq analysis, comparisons that include all these methods are currently lacking. Here, we performed a comprehensive comparison of 18 methods for the identification of DS changes between conditions from multisubject scRNA-seq data. Our results suggest that the pseudobulk methods performed generally best. Both pseudobulks and mixed models that model the subjects as a random effect were superior compared with the naïve single-cell methods that do not model the subjects in any way. While the naïve models achieved higher sensitivity than the pseudobulk methods and the mixed models, they were subject to a high number of false positives. In addition, accounting for subjects through latent variable modeling did not improve the performance of the naïve methods.
单细胞 RNA 测序 (scRNA-seq) 使研究人员能够同时定量数千个细胞的转录组,并研究细胞之间的转录组变化。scRNA-seq 数据集越来越多地包含多主体、多条件实验,以研究条件之间的细胞类型特异性差异状态 (DS)。这可以通过首先识别所有主体中的细胞类型,然后在每个细胞类型内的条件之间执行 DS 分析来完成。在存在生物复制之间的变化的情况下,处理细胞统计独立的天真单细胞 DS 分析方法容易出现假阳性,这是一个称为伪复制偏差的问题。虽然已经提出了几种方法来进行多主体 scRNA-seq 分析中的统计检验,但目前缺乏包含所有这些方法的比较。在这里,我们对 18 种方法进行了全面比较,用于从多主体 scRNA-seq 数据中识别条件之间的 DS 变化。我们的结果表明,伪总体方法的性能通常最好。与不采用任何方式对主体建模的天真单细胞方法相比,伪总体和将主体建模为随机效应的混合模型都具有优势。虽然天真模型的敏感性高于伪总体方法和混合模型,但它们容易出现大量假阳性。此外,通过潜在变量建模考虑主体并没有提高天真方法的性能。