Department of Surgery, Cancer Research Building, St. Louis, MO, USA.
Division of Urologic Surgery Washington University, St. Louis, MO, USA.
Autophagy. 2023 Mar;19(3):1000-1025. doi: 10.1080/15548627.2022.2103961. Epub 2022 Jul 27.
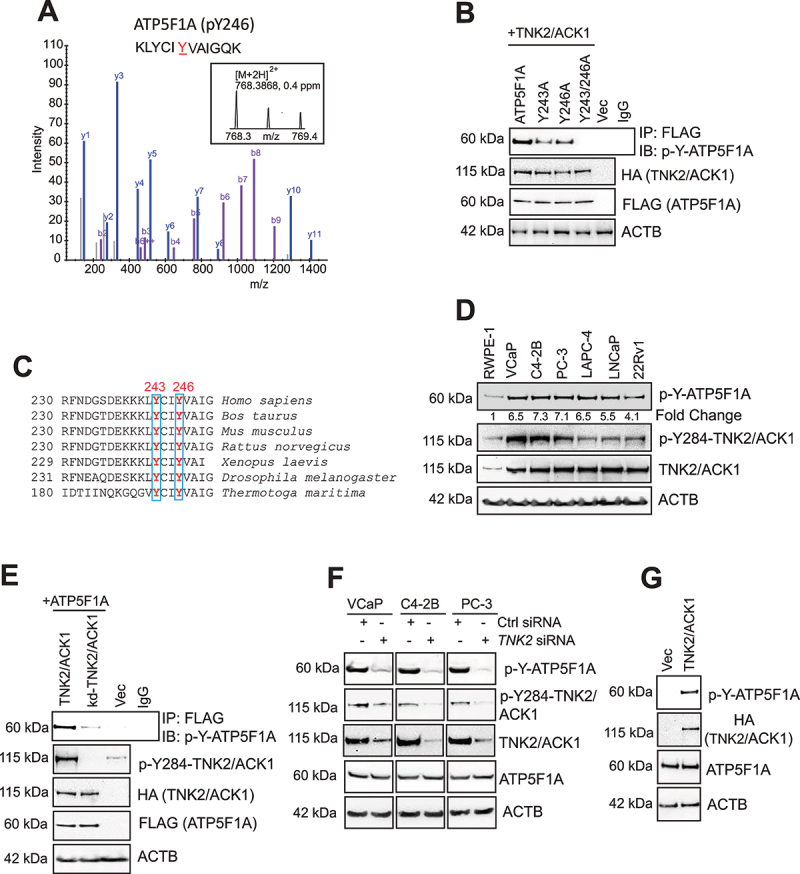
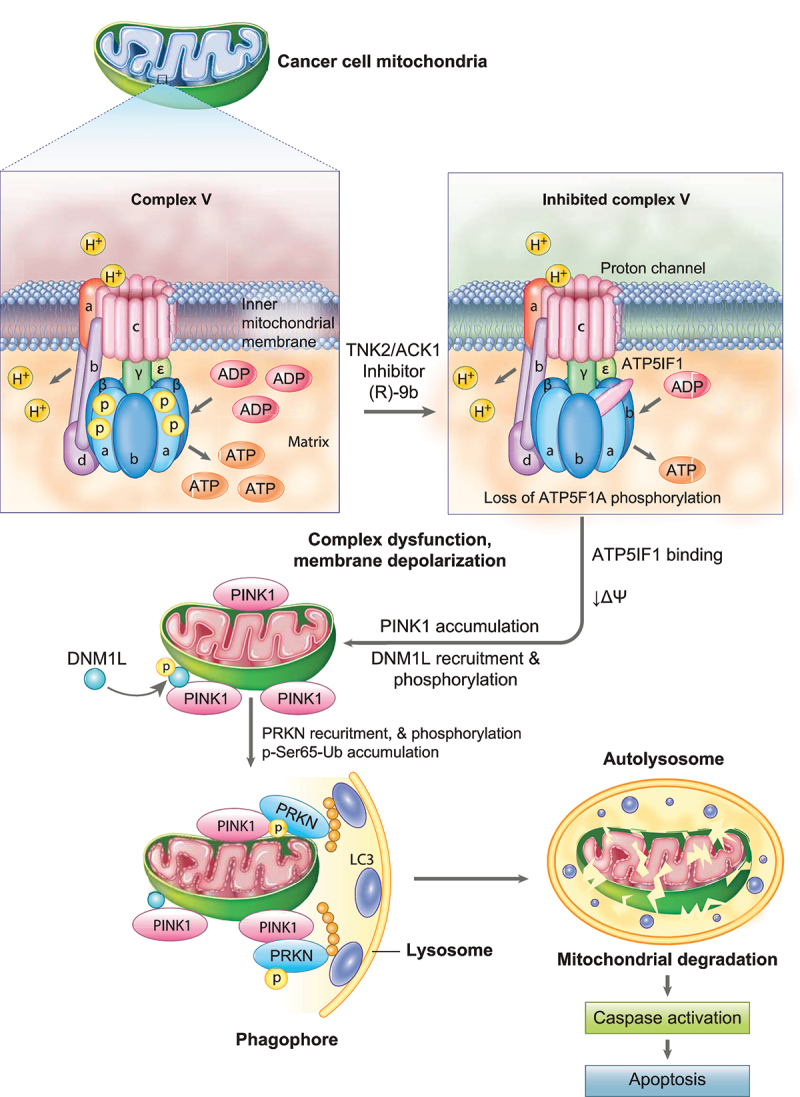
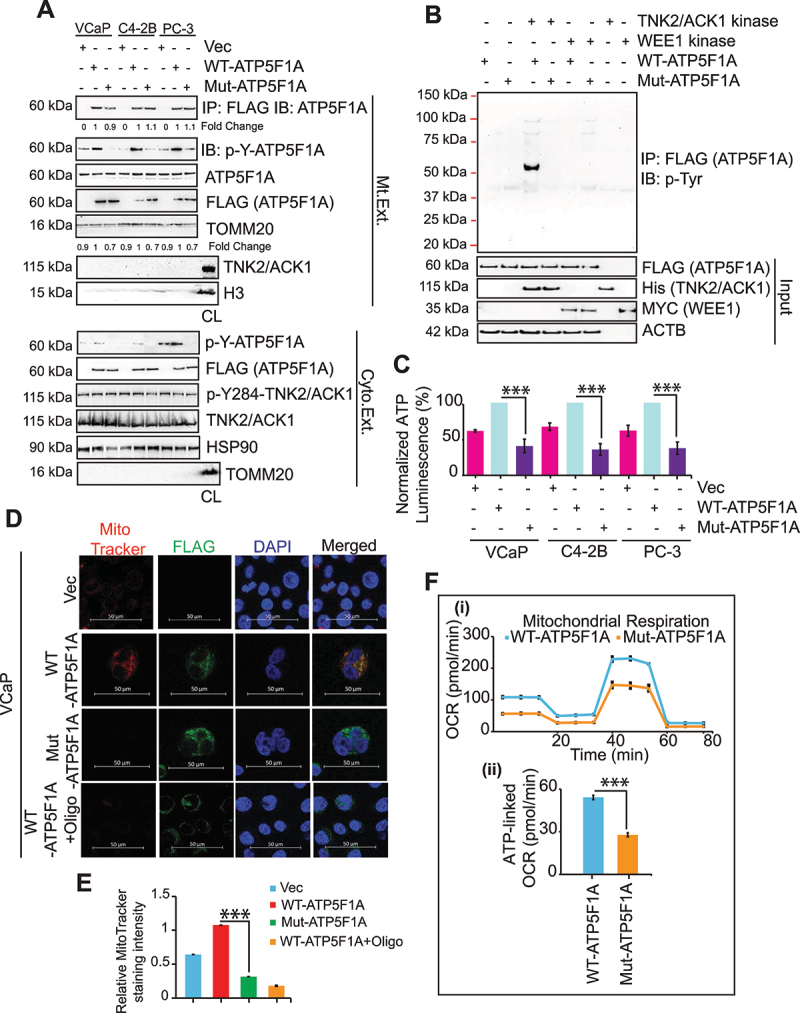
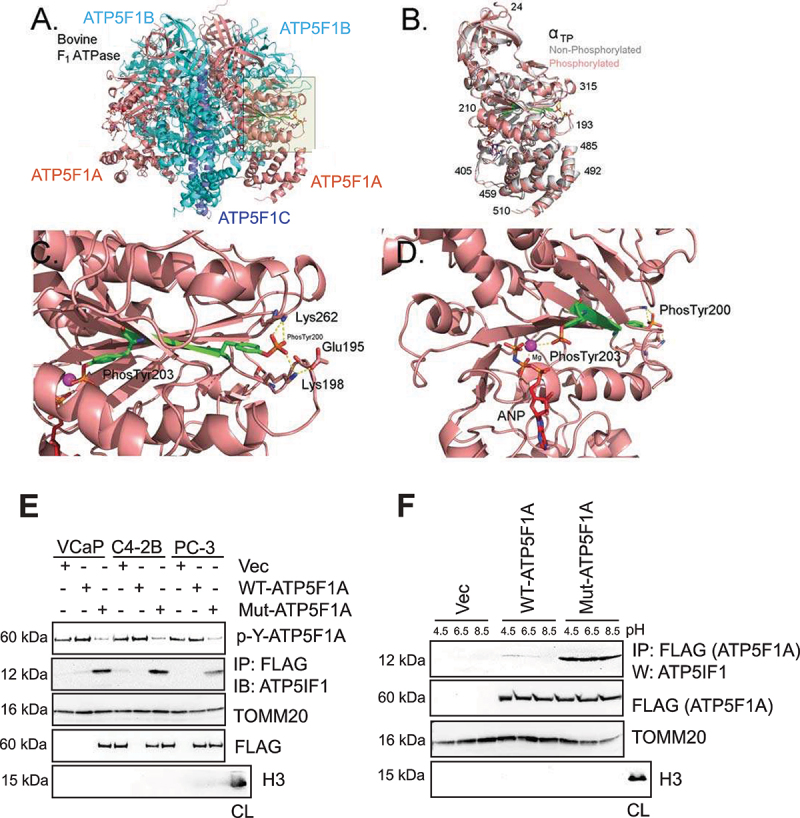
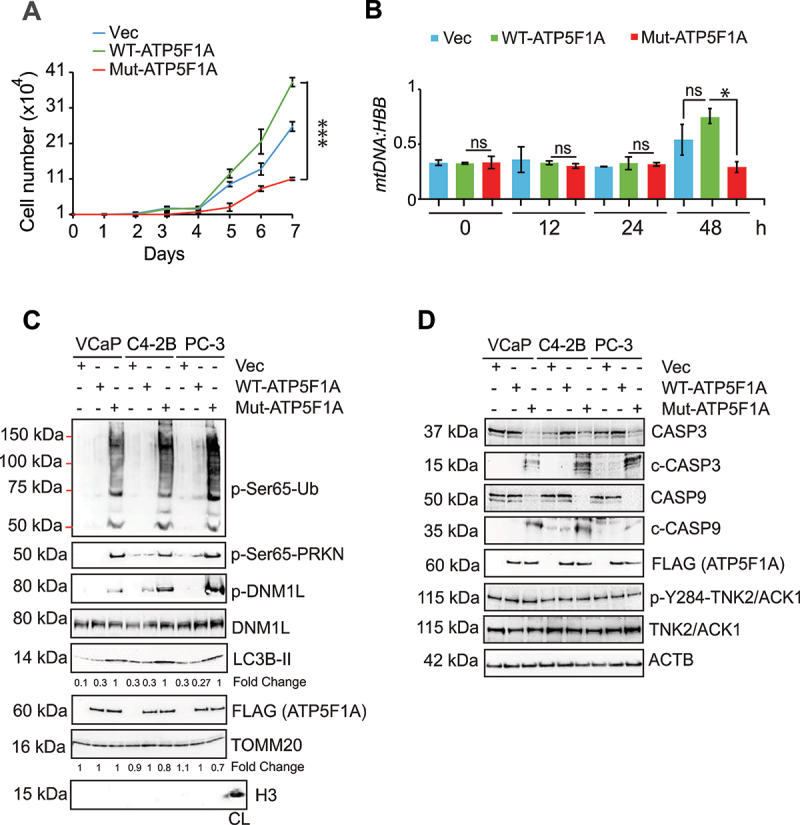
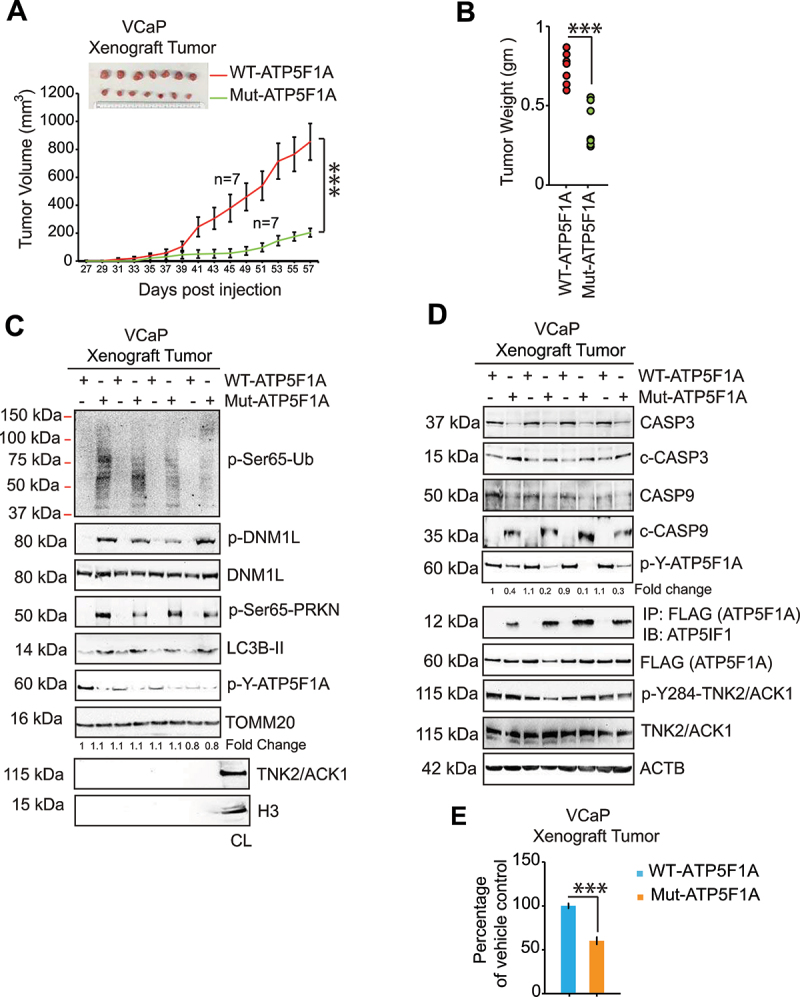
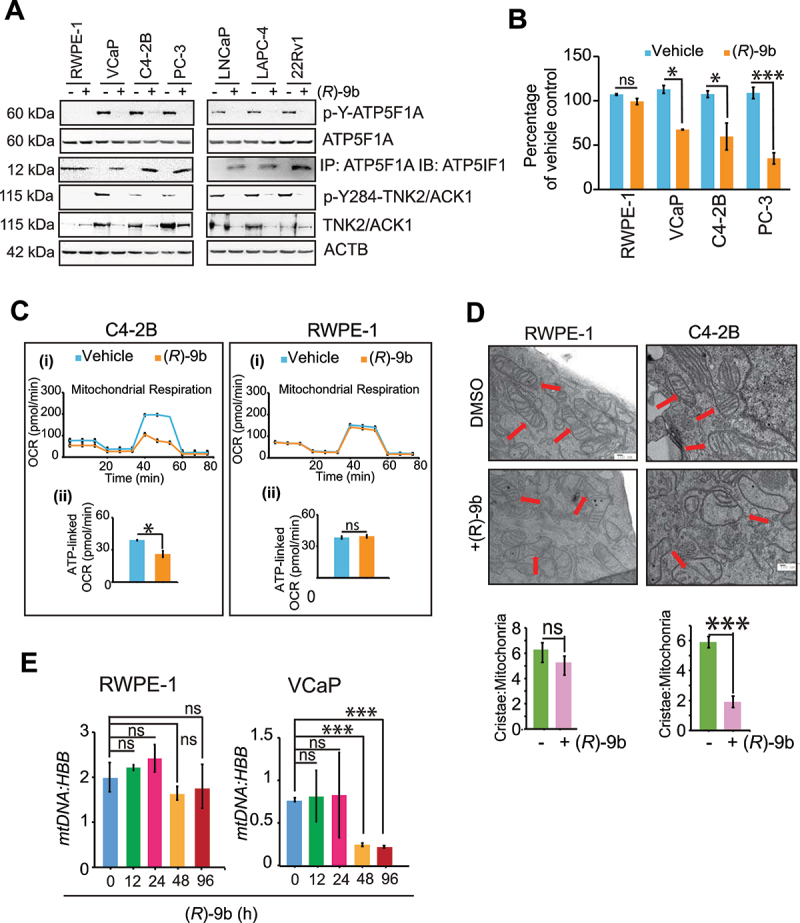
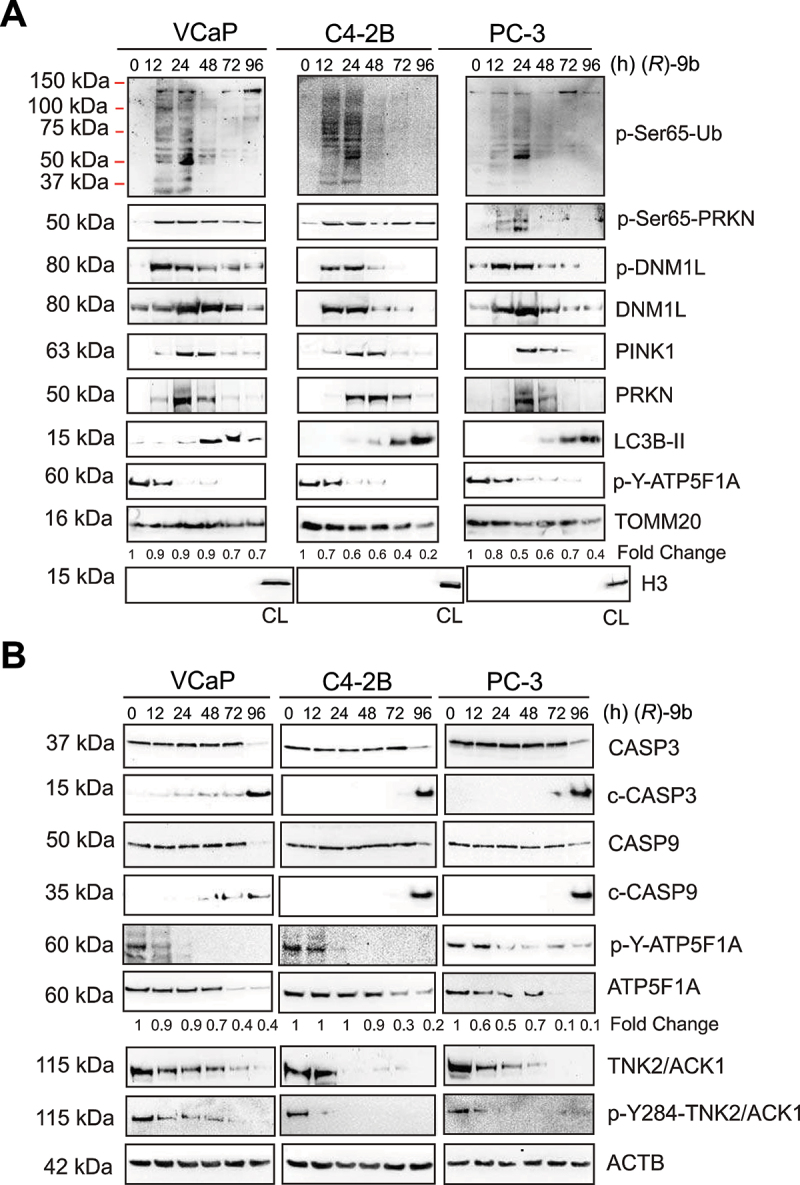
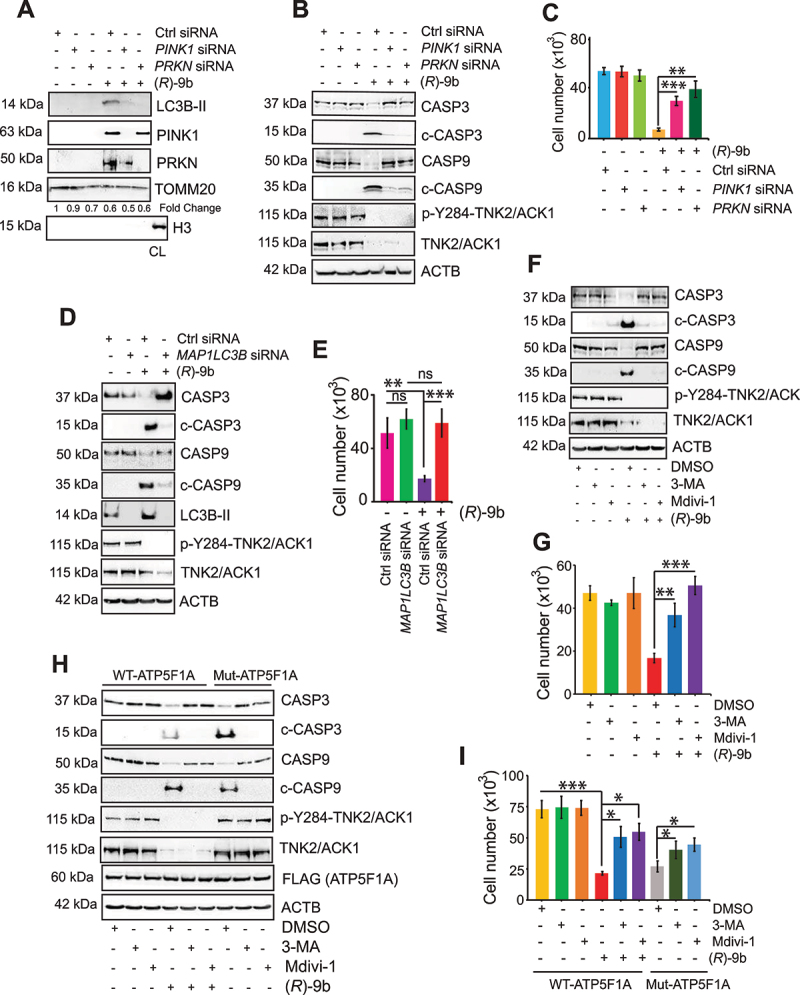
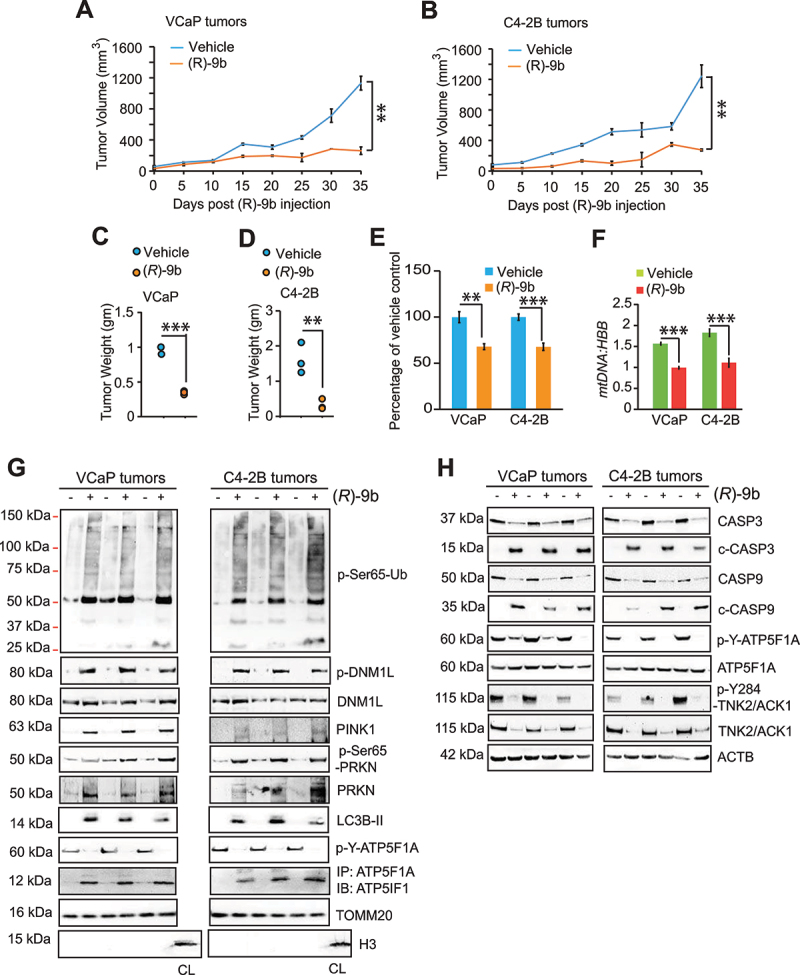
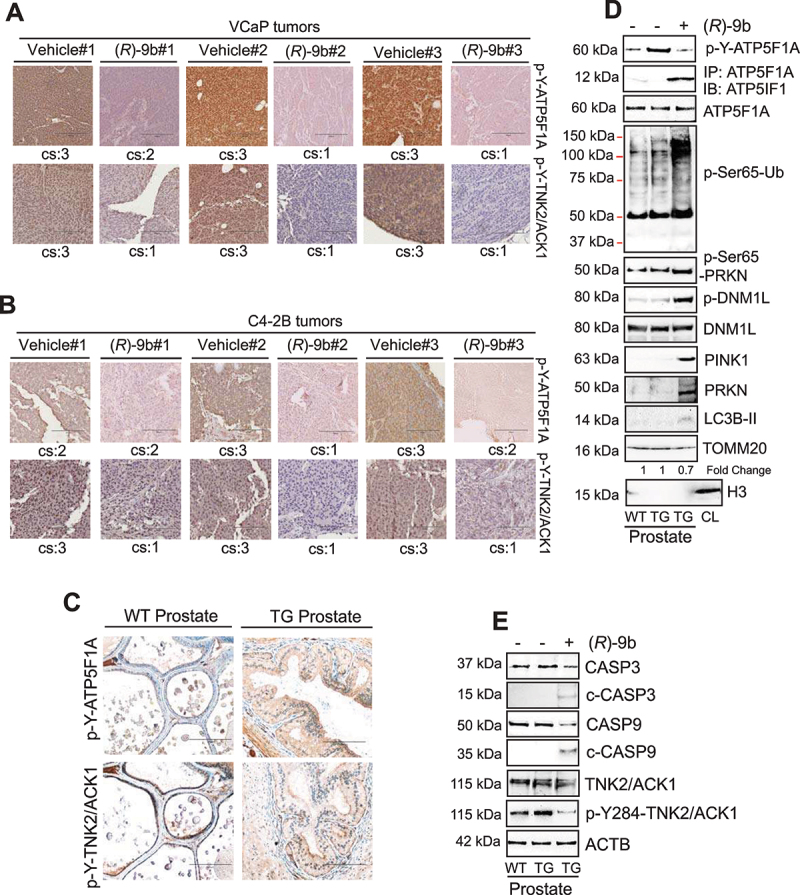
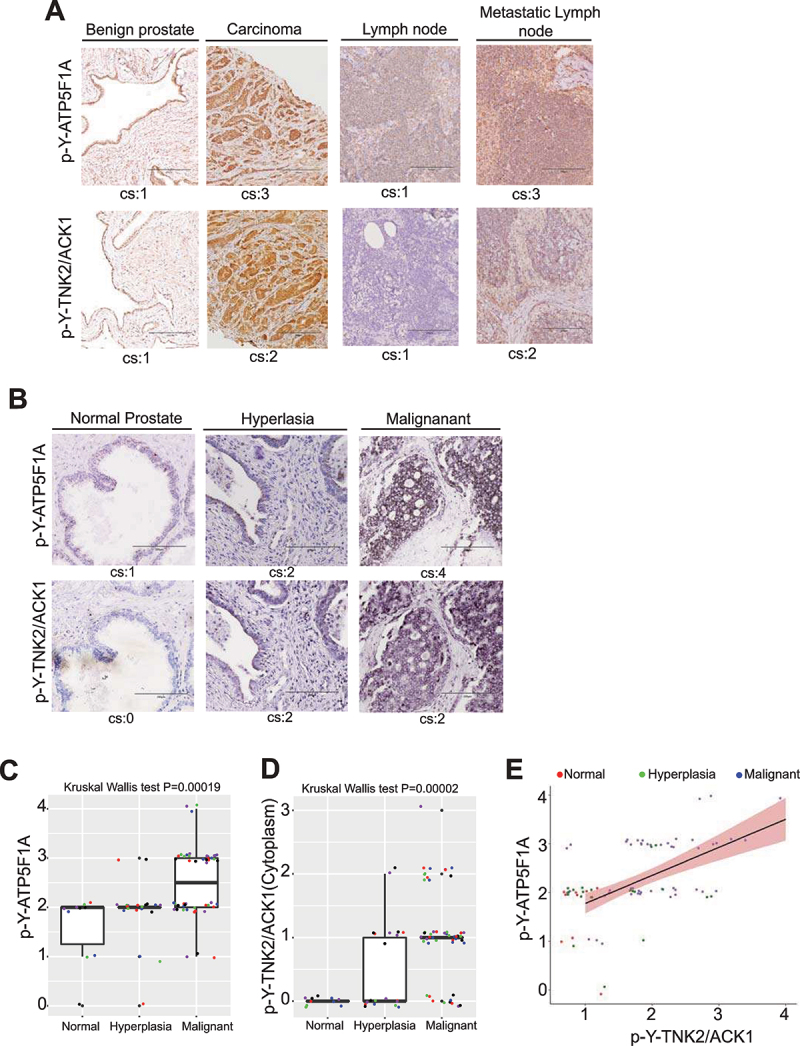
The challenge of rapid macromolecular synthesis enforces the energy-hungry cancer cell mitochondria to switch their metabolic phenotypes, accomplished by activation of oncogenic tyrosine kinases. Precisely how kinase activity is directly exploited by cancer cell mitochondria to meet high-energy demand, remains to be deciphered. Here we show that a non-receptor tyrosine kinase, TNK2/ACK1 (tyrosine kinase non receptor 2), phosphorylated ATP5F1A (ATP synthase F1 subunit alpha) at Tyr243 and Tyr246 (Tyr200 and 203 in the mature protein, respectively) that not only increased the stability of complex V, but also increased mitochondrial energy output in cancer cells. Further, phospho-ATP5F1A (p-Y-ATP5F1A) prevented its binding to its physiological inhibitor, ATP5IF1 (ATP synthase inhibitory factor subunit 1), causing sustained mitochondrial activity to promote cancer cell growth. TNK2 inhibitor, ()- reversed this process and induced mitophagy-based autophagy to mitigate prostate tumor growth while sparing normal prostate cells. Further, depletion of p-Y-ATP5F1A was needed for ()--mediated mitophagic response and tumor growth. Moreover, transgenic mice displayed increased p-Y-ATP5F1A and loss of mitophagy and exhibited formation of prostatic intraepithelial neoplasia (PINs). Consistent with these data, a marked increase in p-Y-ATP5F1A was seen as prostate cancer progressed to the malignant stage. Overall, this study uncovered the molecular intricacy of tyrosine kinase-mediated mitochondrial energy regulation as a distinct cancer cell mitochondrial vulnerability and provided evidence that TNK2 inhibitors can act as "mitocans" to induce cancer-specific mitophagy.: ATP5F1A: ATP synthase F1 subunit alpha; ATP5IF1: ATP synthase inhibitory factor subunit 1; CRPC: castration-resistant prostate cancer; DNM1L: dynamin 1 like; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; Mdivi-1: mitochondrial division inhibitor 1; Mut-ATP5F1A: Y243,246A mutant of ATP5F1A; OXPHOS: oxidative phosphorylation; PC: prostate cancer; PINK1: PTEN induced kinase 1; p-Y-ATP5F1A: phosphorylated tyrosine 243 and 246 on ATP5F1A; TNK2/ACK1: tyrosine kinase non receptor 2; Ub: ubiquitin; WT: wild type.
快速大分子合成的挑战迫使能量饥饿的癌细胞线粒体改变其代谢表型,这是通过激活致癌酪氨酸激酶实现的。激酶活性如何直接被癌细胞线粒体利用来满足高能量需求,仍有待破译。在这里,我们显示一种非受体酪氨酸激酶,TNK2/ACK1(酪氨酸激酶非受体 2),在 Tyr243 和 Tyr246(成熟蛋白中的 Tyr200 和 203)处磷酸化 ATP5F1A(ATP 合酶 F1 亚基 alpha),不仅增加了复合物 V 的稳定性,而且增加了癌细胞中的线粒体能量输出。此外,磷酸化 ATP5F1A(p-Y-ATP5F1A)阻止其与生理抑制剂 ATP5IF1(ATP 合酶抑制因子亚基 1)结合,导致持续的线粒体活性促进癌细胞生长。TNK2 抑制剂()- 逆转了这一过程,并诱导基于噬粒体的自噬来减轻前列腺肿瘤的生长,同时保留正常的前列腺细胞。此外,需要消耗 p-Y-ATP5F1A 来介导噬粒体反应和肿瘤生长。此外,转基因小鼠表现出增加的 p-Y-ATP5F1A 和失去的噬粒体,并表现出前列腺上皮内瘤形成(PINs)的形成。与这些数据一致,在前列腺癌进展为恶性阶段时,p-Y-ATP5F1A 的表达明显增加。总的来说,这项研究揭示了酪氨酸激酶介导的线粒体能量调节的分子复杂性,作为一种独特的癌细胞线粒体脆弱性,并提供了证据表明 TNK2 抑制剂可以作为“mitocans”来诱导癌症特异性噬粒体。