Department of Environmental and Radiological Health Sciences, Colorado State University, Fort Collins, CO 80523, USA.
Molecules. 2022 Jul 25;27(15):4736. doi: 10.3390/molecules27154736.
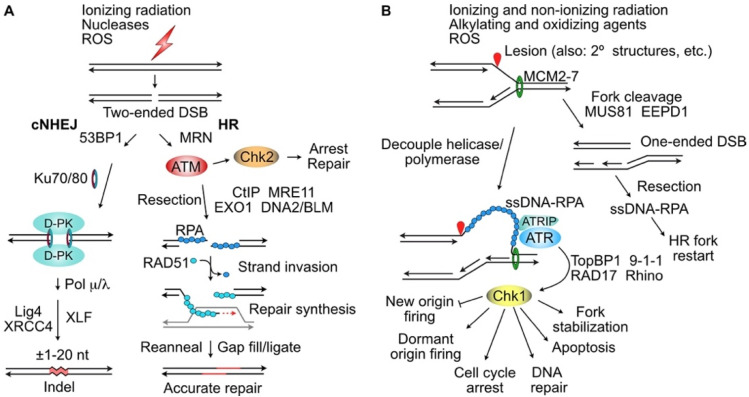
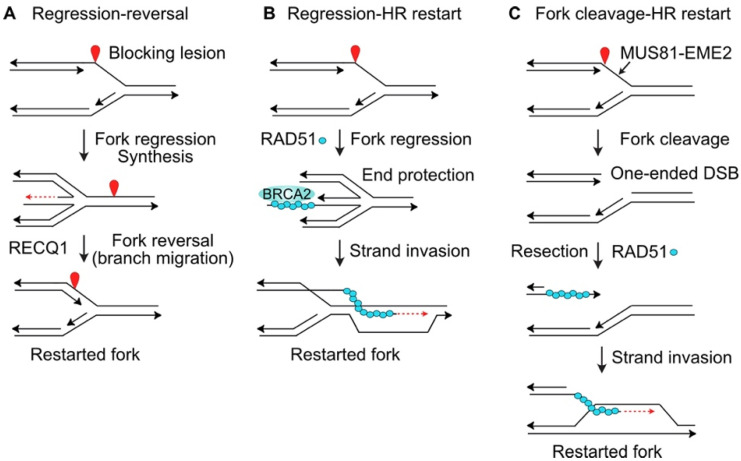
Proliferating cells regularly experience replication stress caused by spontaneous DNA damage that results from endogenous reactive oxygen species (ROS), DNA sequences that can assume secondary and tertiary structures, and collisions between opposing transcription and replication machineries. Cancer cells face additional replication stress, including oncogenic stress that results from the dysregulation of fork progression and origin firing, and from DNA damage induced by radiotherapy and most cancer chemotherapeutic agents. Cells respond to such stress by activating a complex network of sensor, signaling and effector pathways that protect genome integrity. These responses include slowing or stopping active replication forks, protecting stalled replication forks from collapse, preventing late origin replication firing, stimulating DNA repair pathways that promote the repair and restart of stalled or collapsed replication forks, and activating dormant origins to rescue adjacent stressed forks. Currently, most cancer patients are treated with genotoxic chemotherapeutics and/or ionizing radiation, and cancer cells can gain resistance to the resulting replication stress by activating pro-survival replication stress pathways. Thus, there has been substantial effort to develop small molecule inhibitors of key replication stress proteins to enhance tumor cell killing by these agents. Replication stress targets include ATR, the master kinase that regulates both normal replication and replication stress responses; the downstream signaling kinase Chk1; nucleases that process stressed replication forks (MUS81, EEPD1, Metnase); the homologous recombination catalyst RAD51; and other factors including ATM, DNA-PKcs, and PARP1. This review provides an overview of replication stress response pathways and discusses recent pre-clinical studies and clinical trials aimed at improving cancer therapy by targeting replication stress response factors.
增殖细胞经常经历由自发 DNA 损伤引起的复制应激,这些损伤源自内源性活性氧 (ROS)、可以形成二级和三级结构的 DNA 序列,以及转录和复制机器之间的碰撞。癌细胞面临额外的复制应激,包括叉进展和起始点火失调引起的致癌应激,以及放射治疗和大多数癌症化疗药物引起的 DNA 损伤。细胞通过激活保护基因组完整性的传感器、信号和效应途径的复杂网络来应对这种应激。这些反应包括减缓或停止活跃的复制叉,保护停滞的复制叉免受崩溃,防止晚期起始点火,刺激促进停滞或崩溃的复制叉修复和重新启动的 DNA 修复途径,以及激活休眠起始点以挽救相邻受应激的叉。目前,大多数癌症患者接受致基因突变的化疗药物和/或电离辐射治疗,癌细胞可以通过激活生存复制应激途径来获得对由此产生的复制应激的抗性。因此,人们已经做出了巨大的努力来开发关键复制应激蛋白的小分子抑制剂,以增强这些药物对肿瘤细胞的杀伤作用。复制应激靶点包括 ATR,它是调节正常复制和复制应激反应的主激酶;下游信号激酶 Chk1;处理应激复制叉的核酸酶(MUS81、EEPD1、Metnase);同源重组催化剂 RAD51;以及其他因素,包括 ATM、DNA-PKcs 和 PARP1。本文综述了复制应激反应途径,并讨论了最近旨在通过靶向复制应激反应因子改善癌症治疗的临床前研究和临床试验。