Key Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, School of Life Science and Health Engineering, Jiangnan University, 214122, Wuxi, P. R. China.
Department of Medicine, Division of Hematology and Oncology, University of Texas Health, San Antonio, TX, USA.
Cell Death Dis. 2022 Aug 5;13(8):682. doi: 10.1038/s41419-022-05103-1.
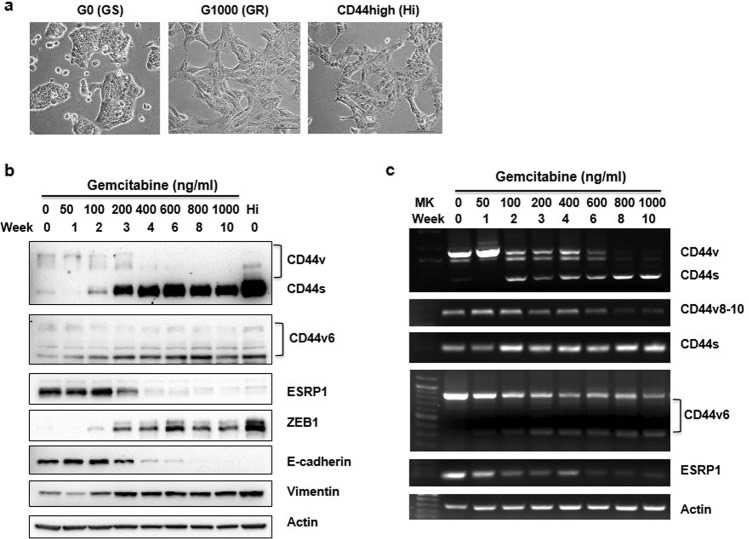
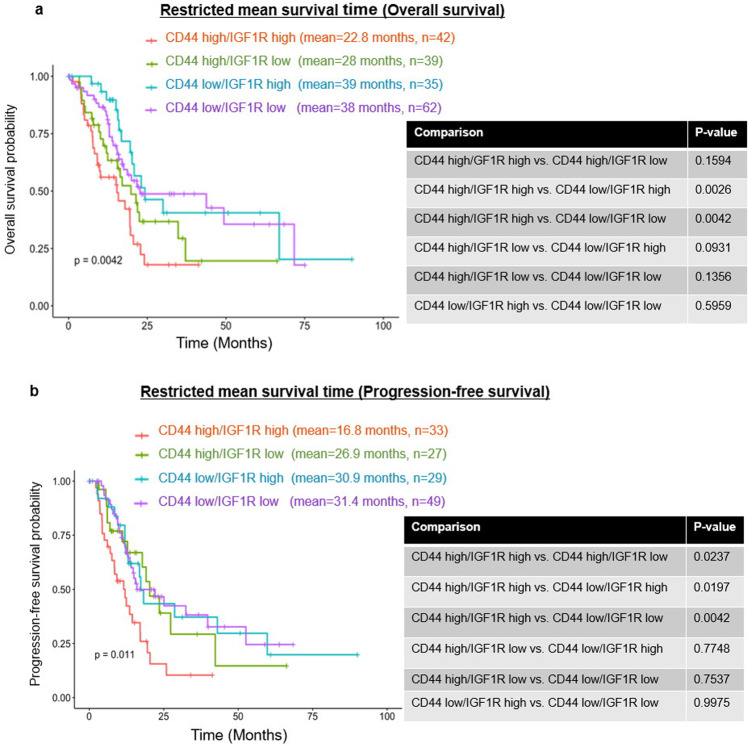
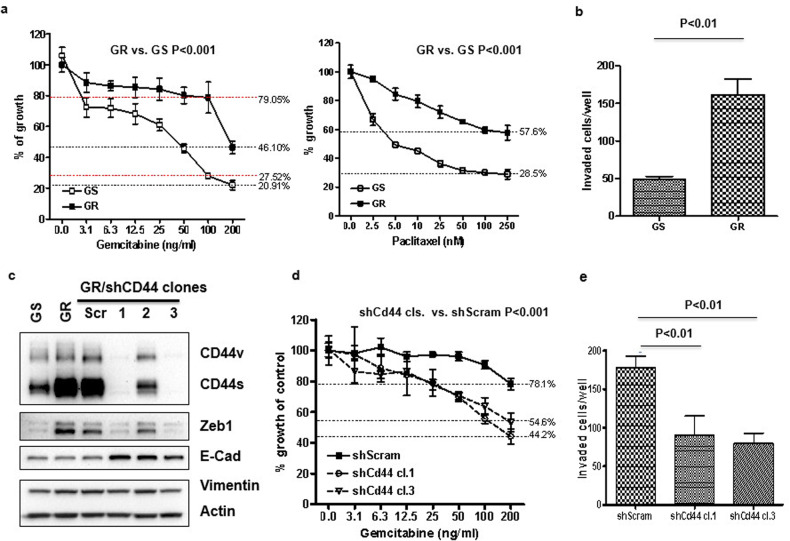
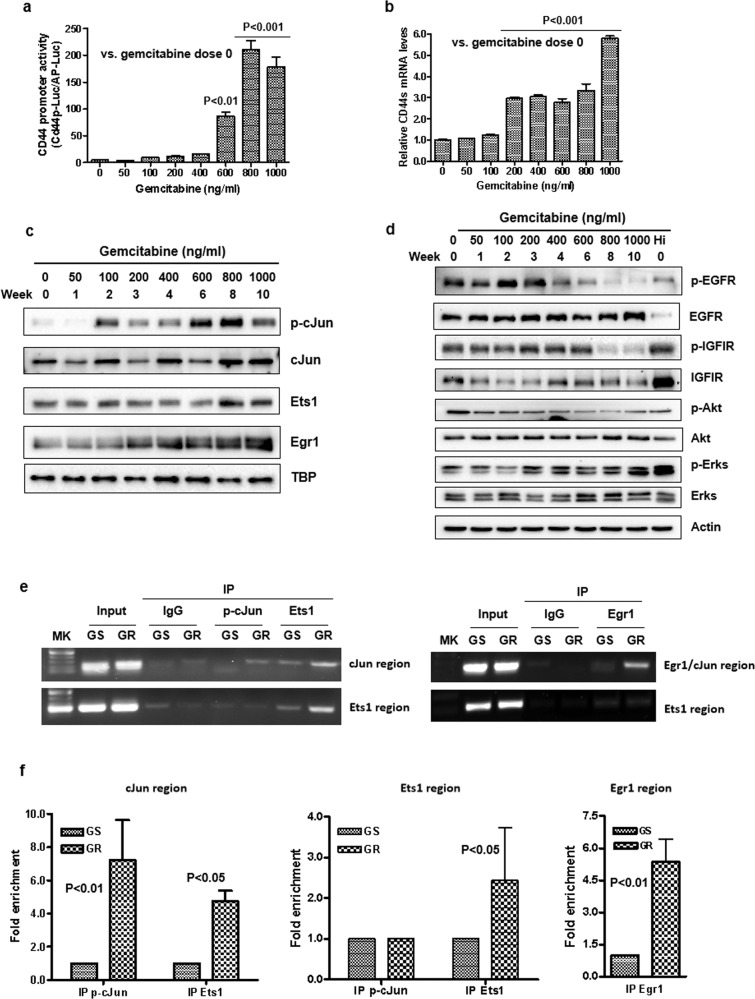
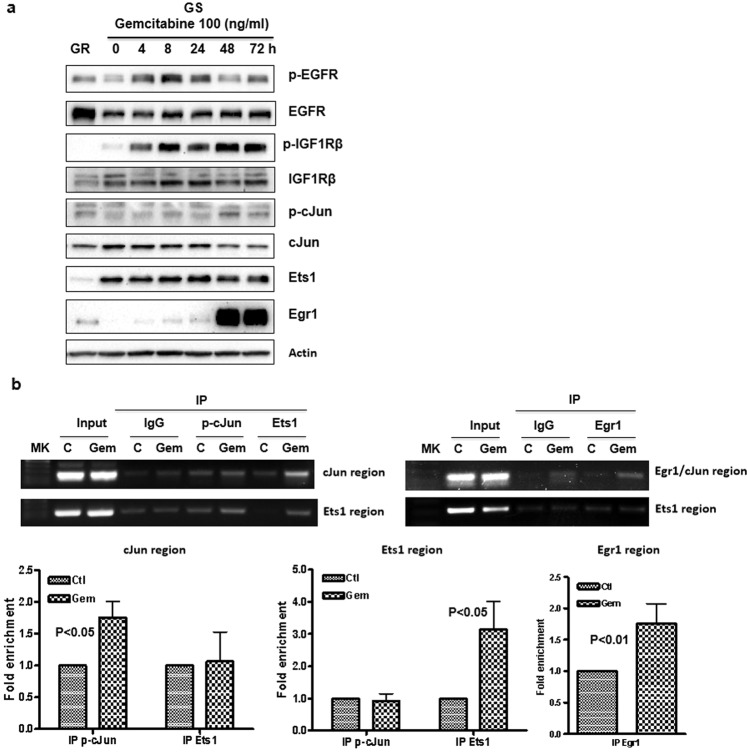
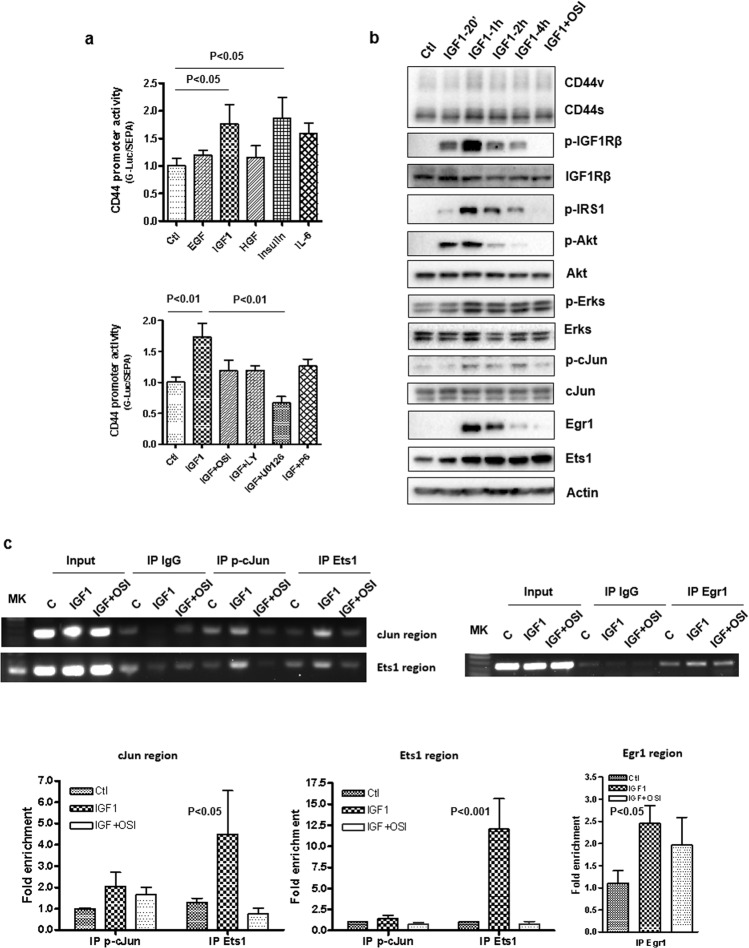
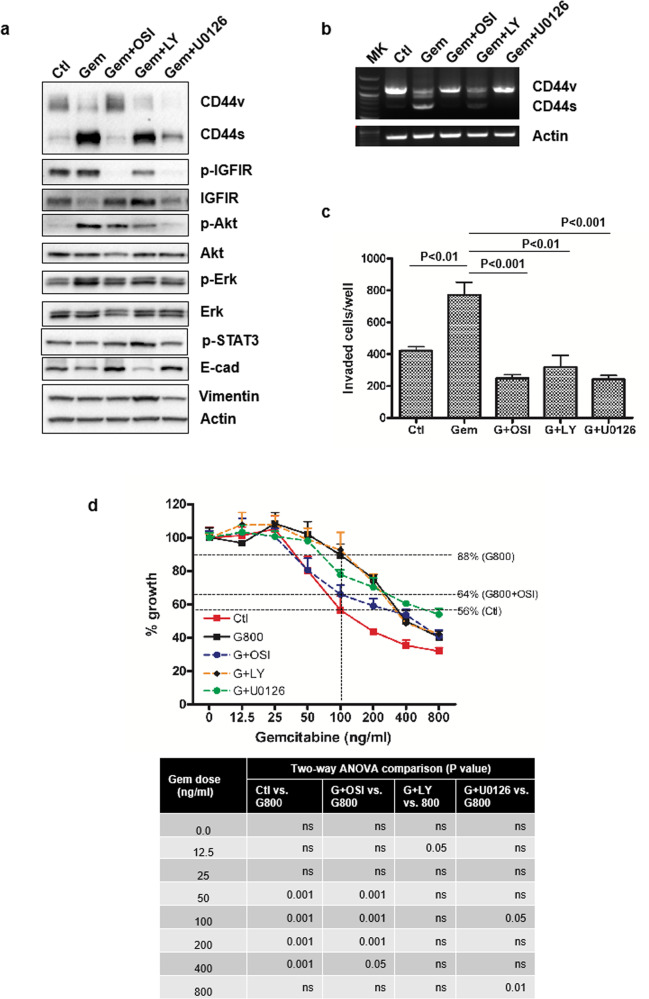
Chemoresistance in pancreatic cancer cells may be caused by the expansion of inherently resistant cancer cells or by the adaptive plasticity of initially sensitive cancer cells. We investigated how CD44 isoforms switching contributed to gemcitabine resistance. Treating CD44 null/low single-cell clones with increasing amounts of gemcitabine caused an increase in expression of CD44 and development of gemcitabine resistant (GR) cells. Drug sensitivity, invasiveness, and EMT process was evaluated by MTT, Matrigel invasion assays, and western blots. Genetic knockdown and pharmacological inhibitors were used to examine the roles of CD44 and IGF1R in mediating gemcitabine resistance. CD44 promoter activity and its interactive EMT-related transcription factors were evaluated by luciferase reporter assay and chromatin immunoprecipitation assay. Kaplan-Meier curve was created by log-rank test to reveal the clinical relevance of CD44 and IGF1R expression in patients. We found silence of CD44 in GR cells partially restored E-cadherin expression, reduced ZEB1 expression, and increased drug sensitivity. The gemcitabine-induced CD44 expressing and isoform switching were associated with an increase in nuclear accumulation of phosphor-cJun, Ets1, and Egr1 and binding of these transcription factors to the CD44 promoter. Gemcitabine treatment induced phosphorylation of IGF1R and increased the expression of phosphor-cJun, Ets1, and Egr1 within 72 h. Stimulation or suppression of IGF1R signaling or its downstream target promoted or blocked CD44 promoter activity. Clinically, patients whose tumors expressed high levels of CD44/IGF1R showed a poor prognosis. This study suggests that IGF1R-dependent CD44 isoform switching confers pancreatic cancer cells to undergo an adaptive change in response to gemcitabine and provides the basis for improved targeted therapy of pancreatic cancer.
胰腺癌细胞的化学抗性可能是由固有抗性癌细胞的扩增或最初敏感癌细胞的适应性可塑性引起的。我们研究了 CD44 异构体转换如何导致吉西他滨耐药。用逐渐增加的吉西他滨处理 CD44 缺失/低单克隆细胞,导致 CD44 的表达增加,并产生吉西他滨耐药(GR)细胞。通过 MTT、Matrigel 侵袭实验和 Western blot 评估药物敏感性、侵袭性和 EMT 过程。使用遗传敲低和药理学抑制剂来研究 CD44 和 IGF1R 在介导吉西他滨耐药中的作用。通过荧光素酶报告实验和染色质免疫沉淀实验评估 CD44 启动子活性及其与 EMT 相关的转录因子的相互作用。通过对数秩检验创建 Kaplan-Meier 曲线,以揭示 CD44 和 IGF1R 在患者中的表达与临床相关性。我们发现,GR 细胞中 CD44 的沉默部分恢复了 E-钙粘蛋白的表达,降低了 ZEB1 的表达,并提高了药物敏感性。吉西他滨诱导的 CD44 表达和异构体转换与核内磷酸化 cJun、Ets1 和 Egr1 的积累以及这些转录因子与 CD44 启动子的结合增加有关。吉西他滨处理诱导 IGF1R 的磷酸化,并在 72 小时内增加磷酸化 cJun、Ets1 和 Egr1 的表达。IGF1R 信号或其下游靶标的刺激或抑制促进或阻断了 CD44 启动子活性。临床上,肿瘤表达高水平 CD44/IGF1R 的患者预后较差。本研究表明,IGF1R 依赖性 CD44 异构体转换赋予胰腺癌细胞对吉西他滨产生适应性变化的能力,并为改善胰腺癌细胞的靶向治疗提供了依据。