Tang Xiaopeng, Zhang Kai, Xiong Kangning
State Engineering Technology Institute for Karst Desertfication Control, School of Karst Science, Guizhou Normal University, Guiyang, China.
College of Animal Science, Shanxi Agricultural University, Jinzhong, China.
Front Vet Sci. 2022 Jul 22;9:894909. doi: 10.3389/fvets.2022.894909. eCollection 2022.
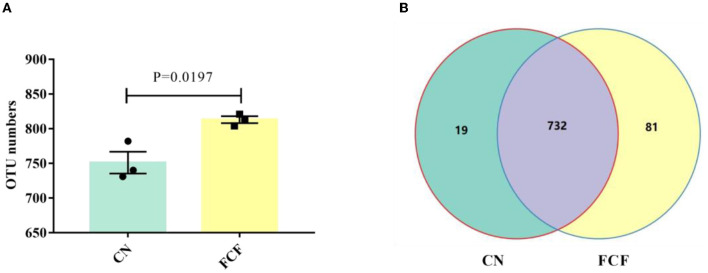
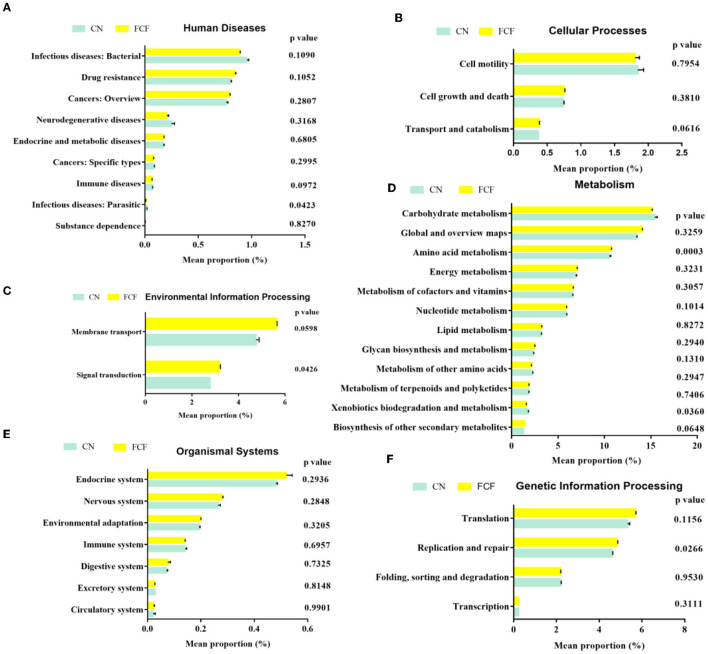
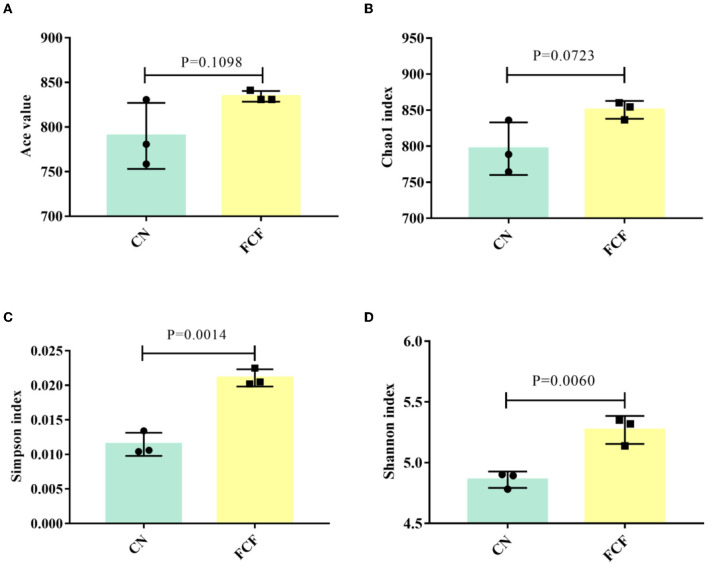
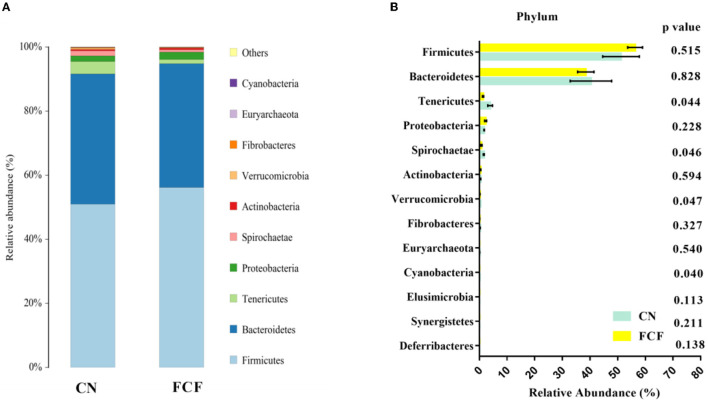
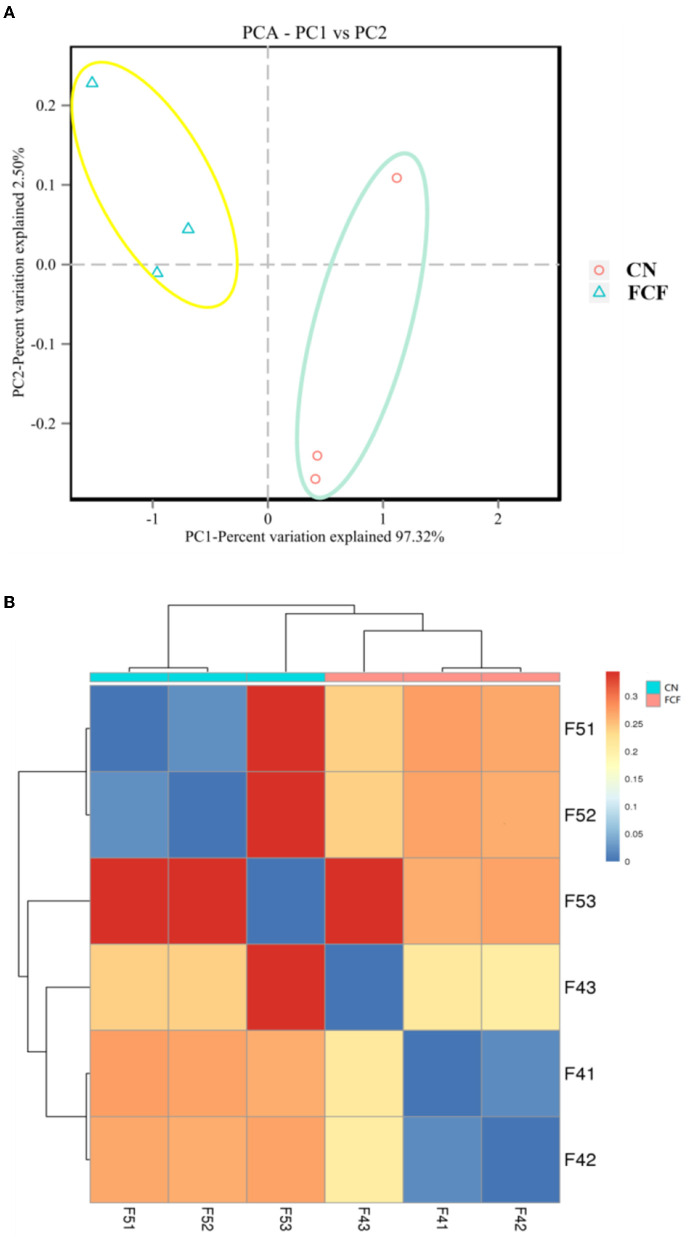
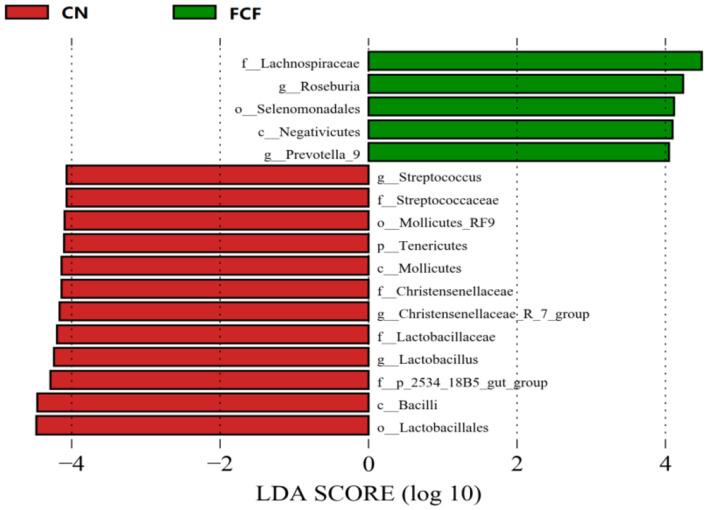
The present study investigated the effects of fermented complete feed (FCF) on fecal microbial composition during the grower-finisher period. A total of 20 pigs (Duroc × Landrace × Yorkshire, 48.74± 1.49 kg) were divided randomly into two groups: the CN group (pigs fed with a basal diet) and the FCF group (pigs fed with FCF). After a 60-day trial period, 3 pigs with middle-weight from each treatment were selected for fecal sampling and fecal microbiota analysis. The results showed that the FCF significantly increased operational taxonomic units (OUT) numbers, alpha diversity (Simpson index and Shannon index), and beta diversity, which means that FCF increased the fecal microbiota diversity. At the phylum level, the abundance of Tenericutes, Spirochaetae, Verrucomicrobia, and Cyanobacteria were changed in pigs fed with FCF; and at the genus level, the abundance of , and were changed in pigs fed with FCF. The linear discriminant analysis effect size (LEfSe) analysis showed that and genera were increased, while Tenericutes phyla and , and genera were decreased in the FCF group compared to the CN group. Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) results predicted that the relative abundance of infectious diseases: parasitic associated genes, xenobiotics biodegradation, and metabolism-associated genes were significantly reduced in the FCF group when compared with the CN group, and the relative abundance of signal transduction associated genes, amino acid metabolism-related genes, and replication and repair associated genes were significantly higher in the FCF group when compared with the CN group. In addition, the relative abundance of transport and catabolism-associated genes, membrane transport-associated genes, and biosynthesis of other secondary metabolite-associated genes tended to be higher in the FCF group when compared with the CN group; and the relative abundance of immune diseases associated genes tended to be lower in the FCF group when compared with the CN group. In conclusion, the FCF influenced the alpha and beta diversity of the fecal microbiota of pigs.
本研究调查了生长育肥期发酵全价饲料(FCF)对猪粪便微生物组成的影响。总共20头猪(杜洛克×长白×大白,体重48.74±1.49千克)被随机分为两组:CN组(饲喂基础日粮的猪)和FCF组(饲喂FCF的猪)。经过60天的试验期后,从每个处理中挑选3头体重中等的猪进行粪便采样和粪便微生物群分析。结果表明,FCF显著增加了可操作分类单元(OUT)数量、α多样性(辛普森指数和香农指数)以及β多样性,这意味着FCF增加了粪便微生物群的多样性。在门水平上,饲喂FCF的猪中柔膜菌门、螺旋体门、疣微菌门和蓝细菌门的丰度发生了变化;在属水平上,饲喂FCF的猪中[此处原文缺失具体属名]的丰度发生了变化。线性判别分析效应大小(LEfSe)分析表明,与CN组相比,FCF组中[此处原文缺失具体属名]属增加,而柔膜菌门以及[此处原文缺失具体属名]属减少。通过未观察状态重建进行群落系统发育研究(PICRUSt)结果预测,与CN组相比,FCF组中传染病:寄生虫相关基因、异生物质生物降解和代谢相关基因的相对丰度显著降低,而信号转导相关基因、氨基酸代谢相关基因以及复制和修复相关基因的相对丰度显著更高。此外,与CN组相比,FCF组中运输和分解代谢相关基因、膜运输相关基因以及其他次生代谢物生物合成相关基因的相对丰度趋于更高;与CN组相比,FCF组中免疫疾病相关基因的相对丰度趋于更低。总之,FCF影响了猪粪便微生物群的α和β多样性。