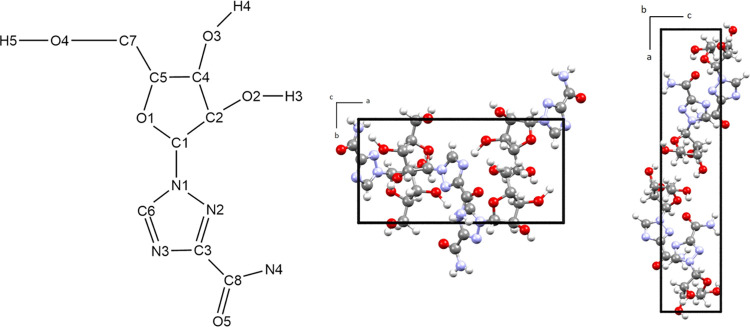
Department of Chemistry, Syracuse University, 1-133 Center for Science and Technology, Syracuse, New York 13244-4100, United States.
Mol Pharm. 2022 Sep 5;19(9):3385-3393. doi: 10.1021/acs.molpharmaceut.2c00509. Epub 2022 Aug 11.
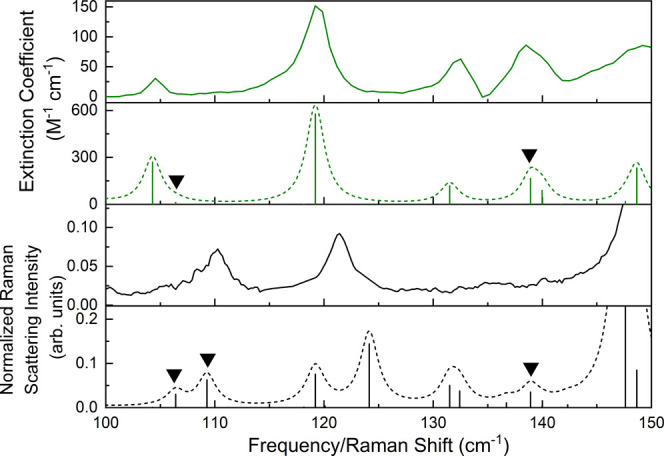
Crystal polymorphism is a common phenomenon in pharmaceutical solids and a critical issue when considering the formulation of therapeutics since multiple polymorphs may form during drug manufacturing. Low-frequency vibrational spectroscopy is sensitive to polymorphic content, and in this work, terahertz time-domain spectroscopy and low-frequency Raman spectroscopy were utilized in the study of crystalline ribavirin, a widely applicable antiviral. Characteristic spectra with numerous peaks in the sub-200 cm region were obtained of the more common polymorph of ribavirin (Form II). Solid-state density functional theory (ss-DFT) simulations were then used to optimize the crystal structure of this polymorph and calculate the frequencies and spectral intensities of the lattice vibrations in the low-frequency region. The near-harmonic thermal behavior of the sample with cooling enabled excellent agreement between experiment and theory to be achieved, emphasizing the quality of the applied model, and the observed spectral peaks could be assigned to specific atomic motions in the solid. Form I and Form II polymorphs of ribavirin were both investigated with ss-DFT to understand the different aspects governing the relative stabilities of these solids. The ss-DFT simulations of the polymorph energies revealed that Form II is more stable at all temperatures due to a stronger cohesive energy than Form I; however, ribavirin in Form I has a significantly lower conformational energy. The finding of monotropism appears to conflict with the reported enantiotropism of the ribavirin polymorphs but ultimately confirms that crystal defects in the real samples greatly affect the thermodynamic relationship of the crystals.
晶体多态性是药物固体中的常见现象,也是考虑治疗药物配方时的一个关键问题,因为在药物制造过程中可能会形成多种多晶型物。低频振动光谱对多晶型物含量敏感,在这项工作中,太赫兹时域光谱和低频拉曼光谱被用于研究广泛应用的抗病毒药物利巴韦林的晶体。获得了利巴韦林(Form II)更常见多晶型物的亚 200cm 区域的特征光谱,其中有许多峰。然后,使用固态密度泛函理论(ss-DFT)模拟来优化该多晶型物的晶体结构,并计算低频区晶格振动的频率和光谱强度。通过冷却实现的样品近谐热行为使得实验和理论之间能够达到极好的一致性,强调了所应用模型的质量,并且可以将观察到的光谱峰分配到固体中的特定原子运动。使用 ss-DFT 研究了利巴韦林的 Form I 和 Form II 多晶型物,以了解控制这些固体相对稳定性的不同方面。多晶型物能量的 ss-DFT 模拟表明,由于比 Form I 更强的内聚能,Form II 在所有温度下都更稳定;然而,Form I 中的利巴韦林具有显著更低的构象能。单态性的发现似乎与报道的利巴韦林多晶型物的反态性相矛盾,但最终证实了实际样品中的晶体缺陷极大地影响了晶体的热力学关系。