Debnath Pinky, Khan Umama, Khan Md Salauddin
Chemical Biotechnology Department, Technical University of Munich, Straubing, Germany.
Biotechnology and Genetic Engineering Discipline, Khulna University, Bangladesh.
Microbiol Insights. 2022 Aug 9;15:11786361221115595. doi: 10.1177/11786361221115595. eCollection 2022.
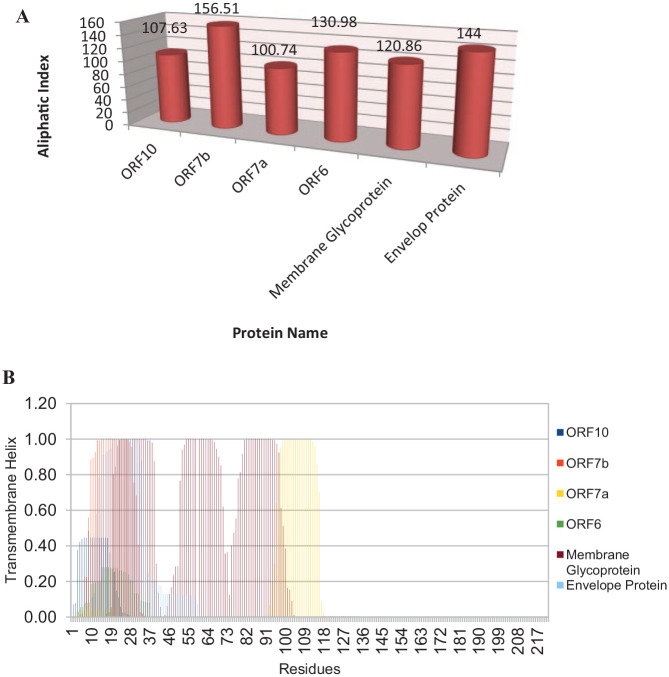
The renowned respiratory disease induced by the severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) has become a global epidemic in just less than a year by the first half of 2020. The subsequent efficient human-to-human transmission of this virus eventually affected millions of people worldwide. The most devastating thing is that the infection rate is continuously uprising and resulting in significant mortality especially among the older age population and those with health co-morbidities. This enveloped, positive-sense RNA virus is chiefly responsible for the infection of the upper respiratory system. The virulence of the SARS-CoV-2 is mostly regulated by its proteins such as entry to the host cell through fusion mechanism, fusion of infected cells with neighboring uninfected cells to spread virus, inhibition of host gene expression, cellular differentiation, apoptosis, mitochondrial biogenesis, etc. But very little is known about the protein structures and functionalities. Therefore, the main purpose of this study is to learn more about these proteins through bioinformatics approaches. In this study, ORF10, ORF7b, ORF7a, ORF6, membrane glycoprotein, and envelope protein have been selected from a Bangladeshi Corona-virus strain G039392 and a number of bioinformatics tools (MEGA-X-V10.1.7, PONDR, ProtScale, ProtParam, SCRIBER, NetSurfP v2.0, IntFOLD, UCSF Chimera, and PyMol) and strategies were implemented for multiple sequence alignment and phylogeny analysis with 9 different variants, predicting hydropathicity, amino acid compositions, protein-binding propensity, protein disorders, and 2D and 3D protein modeling. Selected proteins were characterized as highly flexible, structurally and electrostatically extremely stable, ordered, biologically active, hydrophobic, and closely related to proteins of different variants. This detailed information regarding the characterization and structure of proteins of SARS-CoV-2 Bangladeshi variant was performed for the first time ever to unveil the deep mechanism behind the virulence features. And this robust appraisal also paves the future way for molecular docking, vaccine development targeting these characterized proteins.
由严重急性呼吸综合征冠状病毒2(SARS-CoV-2)引起的著名呼吸道疾病在2020年上半年不到一年的时间里就已成为全球流行病。这种病毒随后高效的人际传播最终影响了全球数百万人。最具破坏性的是,感染率持续上升,导致大量死亡,尤其是在老年人群和有健康合并症的人群中。这种包膜的正链RNA病毒主要负责上呼吸道感染。SARS-CoV-2的毒力主要由其蛋白质调节,例如通过融合机制进入宿主细胞、感染细胞与相邻未感染细胞融合以传播病毒、抑制宿主基因表达、细胞分化、凋亡、线粒体生物发生等。但对其蛋白质结构和功能知之甚少。因此,本研究的主要目的是通过生物信息学方法更多地了解这些蛋白质。在本研究中,从孟加拉国冠状病毒株G039392中选择了ORF10、ORF7b、ORF7a、ORF6、膜糖蛋白和包膜蛋白,并使用了多种生物信息学工具(MEGA-X-V10.1.7、PONDR、ProtScale、ProtParam、SCRIBER、NetSurfP v2.0、IntFOLD、UCSF Chimera和PyMol),实施了与9种不同变体进行多序列比对和系统发育分析、预测亲水性、氨基酸组成、蛋白质结合倾向、蛋白质无序性以及二维和三维蛋白质建模的策略。所选蛋白质的特征为高度灵活、结构和静电极其稳定、有序、具有生物活性、疏水,并且与不同变体的蛋白质密切相关。有史以来首次对SARS-CoV-2孟加拉国变体蛋白质的特征和结构进行了详细信息分析,以揭示毒力特征背后的深层机制。这种有力的评估也为分子对接、针对这些已表征蛋白质的疫苗开发铺平了未来道路。