Biozentrum, Universität Basel, Basel, Switzerland.
Swiss Institute of Bioinformatics, Basel, Switzerland.
PLoS Comput Biol. 2022 Aug 19;18(8):e1010394. doi: 10.1371/journal.pcbi.1010394. eCollection 2022 Aug.
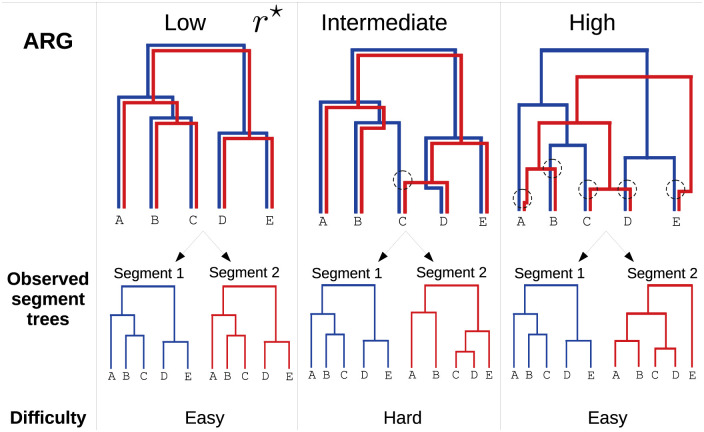
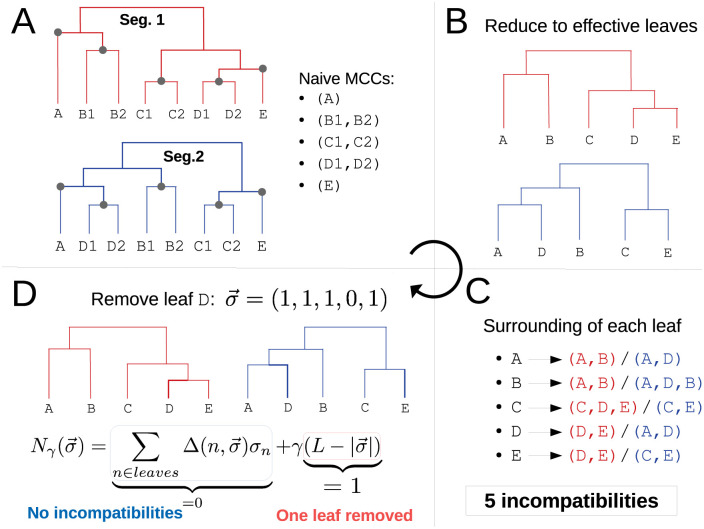
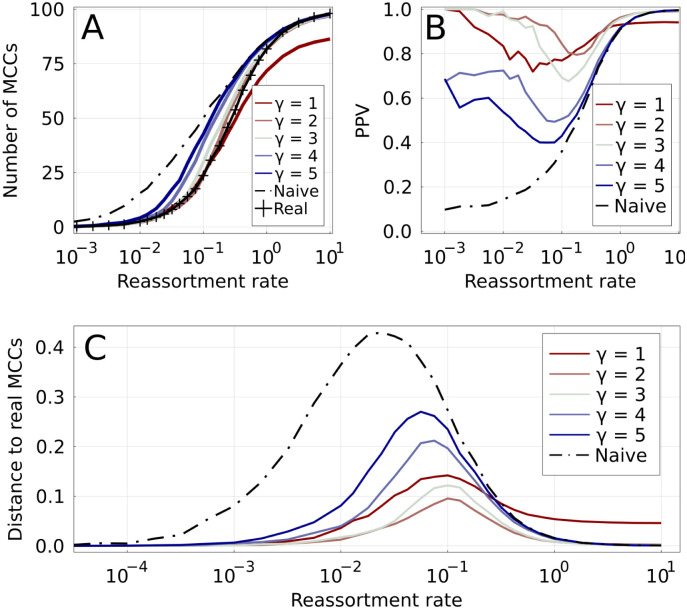
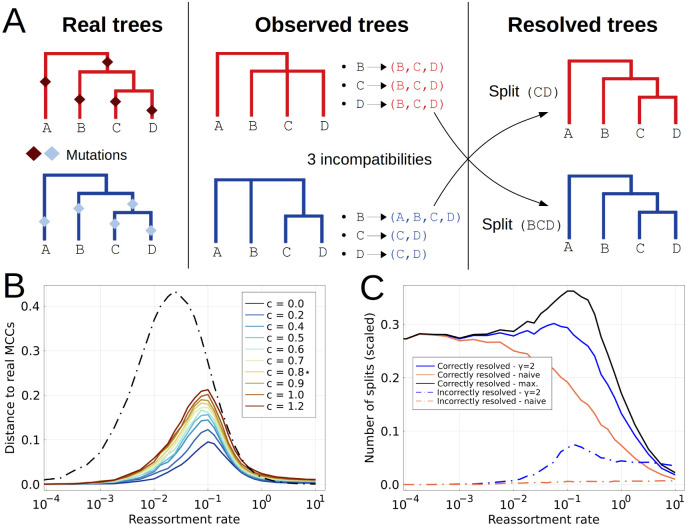
When two influenza viruses co-infect the same cell, they can exchange genome segments in a process known as reassortment. Reassortment is an important source of genetic diversity and is known to have been involved in the emergence of most pandemic influenza strains. However, because of the difficulty in identifying reassortment events from viral sequence data, little is known about their role in the evolution of the seasonal influenza viruses. Here we introduce TreeKnit, a method that infers ancestral reassortment graphs (ARG) from two segment trees. It is based on topological differences between trees, and proceeds in a greedy fashion by finding regions that are compatible in the two trees. Using simulated genealogies with reassortments, we show that TreeKnit performs well in a wide range of settings and that it is as accurate as a more principled bayesian method, while being orders of magnitude faster. Finally, we show that it is possible to use the inferred ARG to better resolve segment trees and to construct more informative visualizations of reassortments.
当两种流感病毒同时感染同一细胞时,它们可以在称为重配的过程中交换基因组片段。重配是遗传多样性的重要来源,已知与大多数大流行性流感毒株的出现有关。但是,由于从病毒序列数据中识别重配事件很困难,因此人们对其在季节性流感病毒进化中的作用知之甚少。在这里,我们介绍了一种从两个片段树推断祖先重配图(ARG)的方法TreeKnit。它基于树之间的拓扑差异,并通过在两棵树中找到兼容的区域以贪婪的方式进行操作。使用带有重配的模拟系统发育树,我们表明 TreeKnit 在广泛的设置中表现良好,并且与更有原则的贝叶斯方法一样准确,而速度则快几个数量级。最后,我们表明,可以使用推断出的 ARG 更好地解析片段树,并构建更具信息量的重配可视化效果。