Liu Miaosen, Yang Jian, Duan Huilong, Yu Lan, Wu Dingwen, Li Haomin
School of Medicine, Zhejiang University, Hangzhou, China.
The College of Biomedical Engineering and Instrument Science, Zhejiang University, Hangzhou, China.
Front Genet. 2022 Aug 19;13:985500. doi: 10.3389/fgene.2022.985500. eCollection 2022.
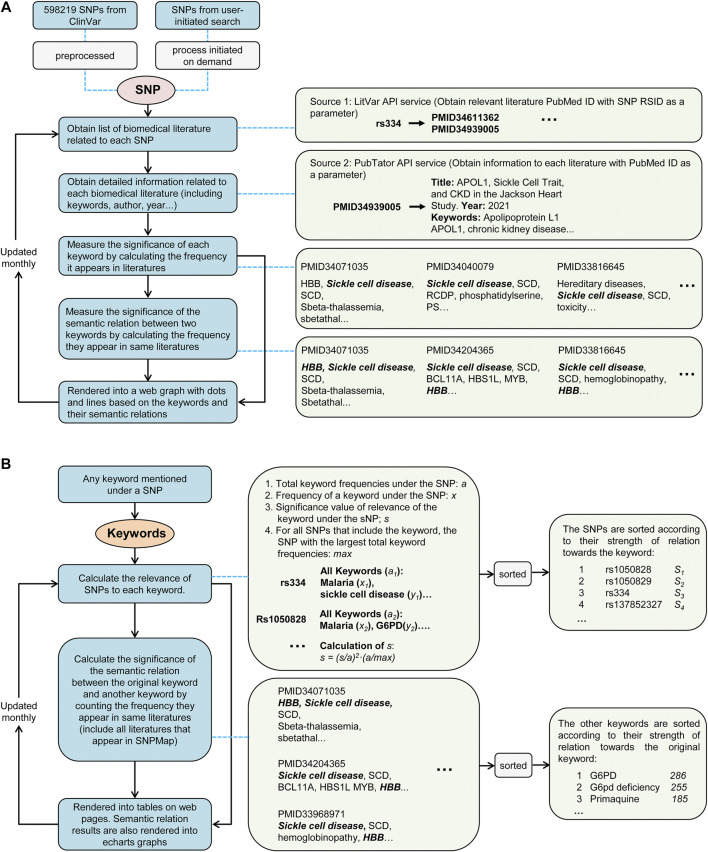
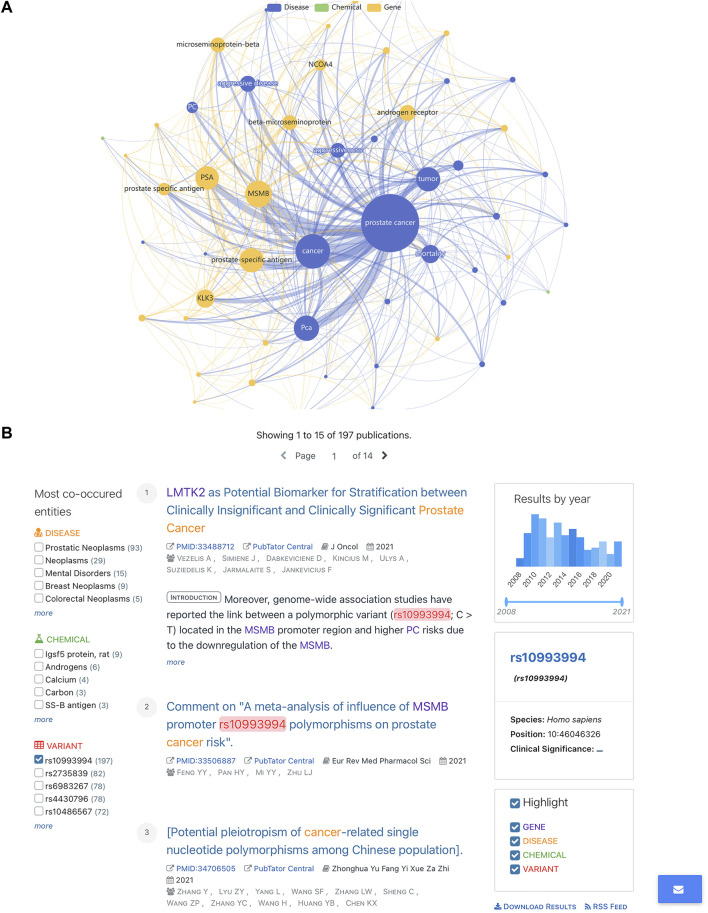
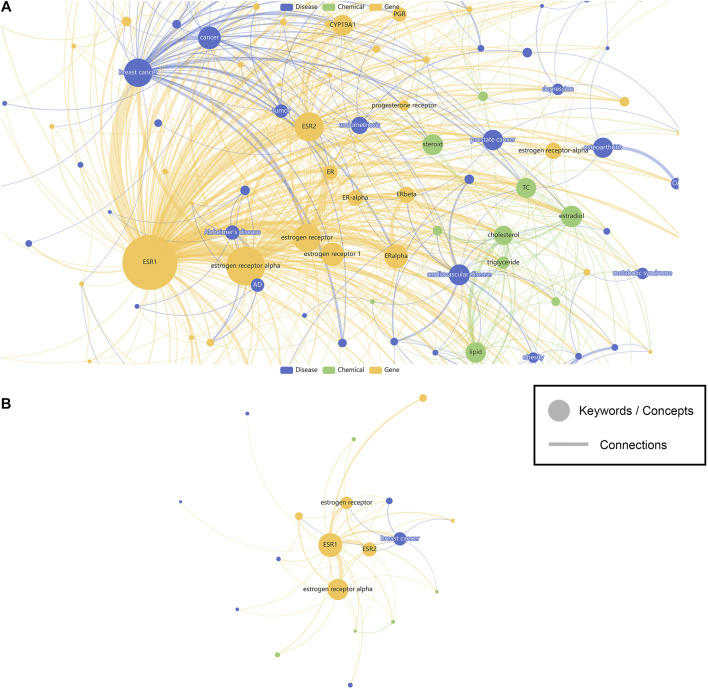
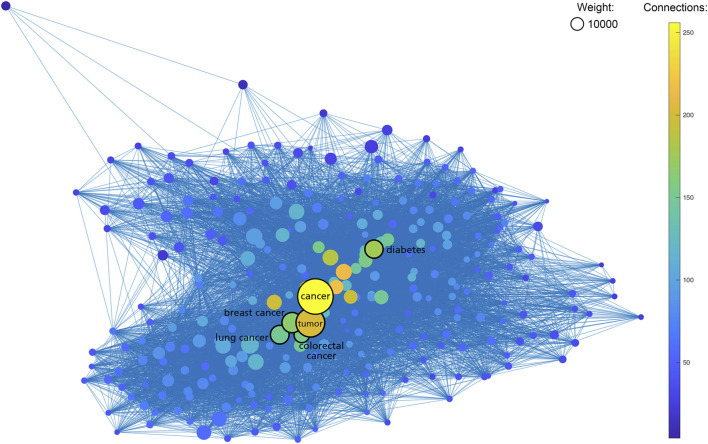
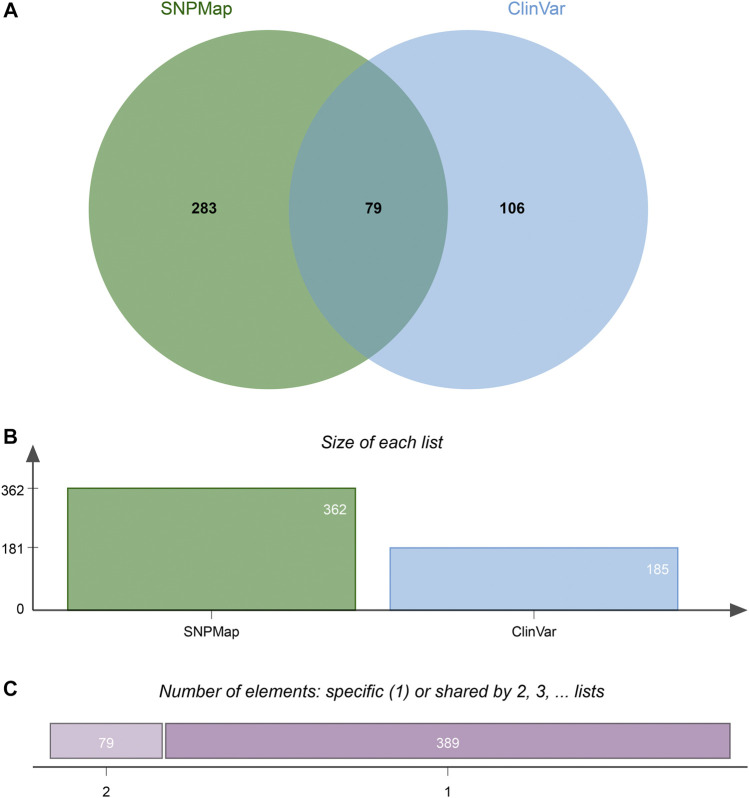
New technologies, such as next-generation sequencing, have advanced the ability to diagnose diseases and improve prognosis but require the identification of thousands of variants in each report based on several databases scattered across places. Curating an integrated interpretation database is time-consuming, costly, and needs regular update. On the other hand, the automatic curation of knowledge sources always results in overloaded information. In this study, an automated pipeline was proposed to create an integrated visual single-nucleotide polymorphism (SNP) interpretation tool called SNPMap. SNPMap pipelines periodically obtained SNP-related information from LitVar, PubTator, and GWAS Catalog API tools and presented it to the user after extraction, integration, and visualization. Keywords and their semantic relations to each SNP are rendered into two graphs, with their significance represented by the size/width of circles/lines. Moreover, the most related SNPs for each keyword that appeared in SNPMap were calculated and sorted. SNPMap retains the advantage of an automatic process while assisting users in accessing more lucid and detailed information through visualization and integration with other materials.
诸如新一代测序等新技术提升了疾病诊断能力并改善了预后,但基于分散在各地的多个数据库,每份报告都需要识别数千个变异。构建一个综合解读数据库既耗时又昂贵,还需要定期更新。另一方面,知识源的自动整理总会导致信息过载。在本研究中,我们提出了一个自动化流程来创建一个名为SNPMap的综合可视化单核苷酸多态性(SNP)解读工具。SNPMap流程会定期从LitVar、PubTator和GWAS Catalog API工具中获取与SNP相关的信息,并在提取、整合和可视化后呈现给用户。关键词及其与每个SNP的语义关系被绘制成两个图表,其重要性由圆圈/线条的大小/宽度表示。此外,还计算并排序了在SNPMap中出现的每个关键词的最相关SNP。SNPMap保留了自动化流程的优势,同时通过可视化以及与其他材料的整合,帮助用户获取更清晰、详细的信息。