Centre for Computational Natural Sciences and Bioinformatics, International Institute of Information Technology, Hyderabad, 500032, India.
UM-DAE-Centre For Excellence In Basic Sciences, University of Mumbai, Vidyanagari, Mumbai, India.
Sci Data. 2022 Sep 7;9(1):548. doi: 10.1038/s41597-022-01631-9.
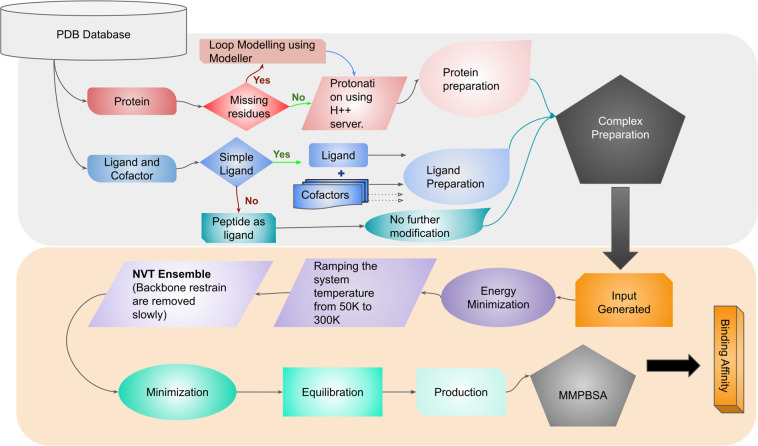
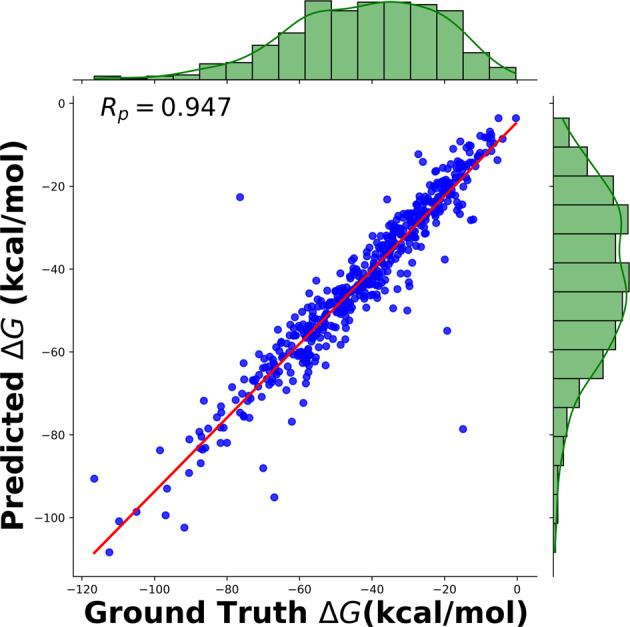
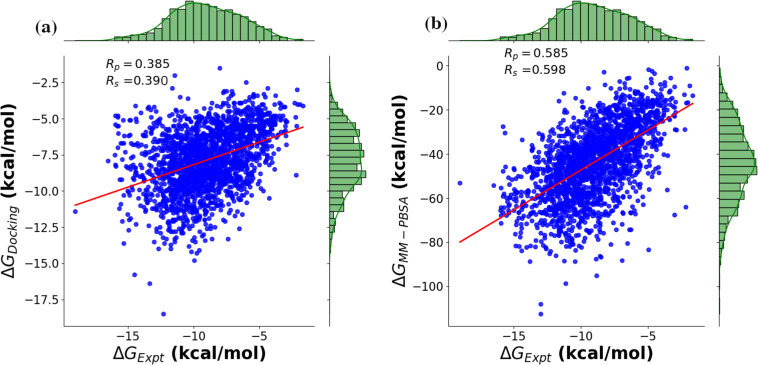
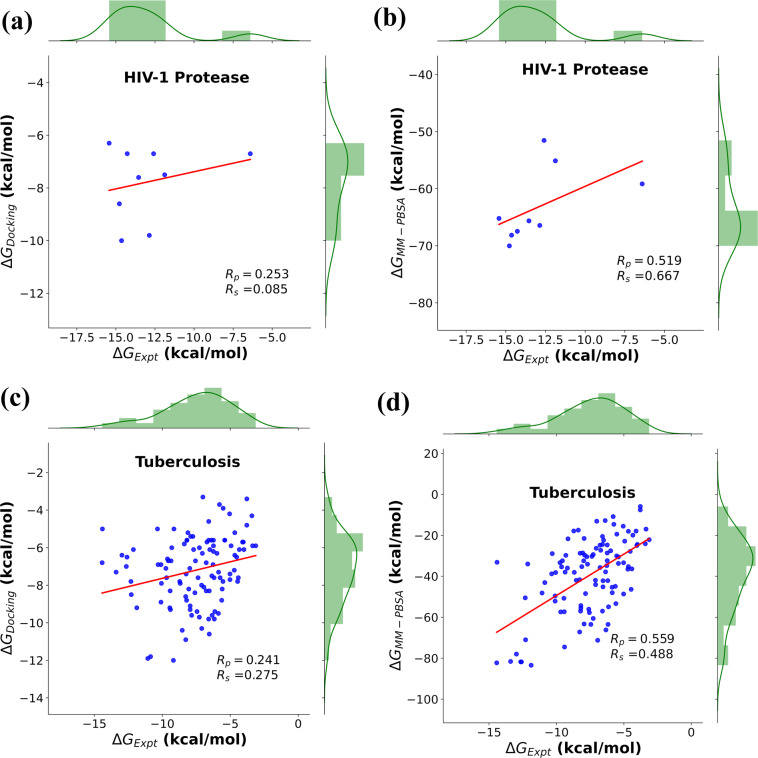
Computational methods and recently modern machine learning methods have played a key role in structure-based drug design. Though several benchmarking datasets are available for machine learning applications in virtual screening, accurate prediction of binding affinity for a protein-ligand complex remains a major challenge. New datasets that allow for the development of models for predicting binding affinities better than the state-of-the-art scoring functions are important. For the first time, we have developed a dataset, PLAS-5k comprised of 5000 protein-ligand complexes chosen from PDB database. The dataset consists of binding affinities along with energy components like electrostatic, van der Waals, polar and non-polar solvation energy calculated from molecular dynamics simulations using MMPBSA (Molecular Mechanics Poisson-Boltzmann Surface Area) method. The calculated binding affinities outperformed docking scores and showed a good correlation with the available experimental values. The availability of energy components may enable optimization of desired components during machine learning-based drug design. Further, OnionNet model has been retrained on PLAS-5k dataset and is provided as a baseline for the prediction of binding affinities.
计算方法和最近的机器学习方法在基于结构的药物设计中发挥了关键作用。尽管有几个基准数据集可用于虚拟筛选中的机器学习应用,但准确预测蛋白质-配体复合物的结合亲和力仍然是一个主要挑战。新的数据集对于开发能够比最新评分函数更好地预测结合亲和力的模型非常重要。我们首次开发了一个数据集 PLAS-5k,其中包含了从 PDB 数据库中选择的 5000 个蛋白质-配体复合物。该数据集包含结合亲和力以及能量成分,如静电、范德华、极性和非极性溶剂化能,这些能量成分是使用 MMPBSA(分子力学泊松-玻尔兹曼表面积)方法从分子动力学模拟中计算得出的。计算出的结合亲和力优于对接评分,并与可用的实验值显示出良好的相关性。能量成分的可用性可能使在基于机器学习的药物设计中能够优化所需的成分。此外,OnionNet 模型已经在 PLAS-5k 数据集上进行了重新训练,并作为预测结合亲和力的基线提供。