Molecular Targets and Therapeutics Center, Institute of Structural Biology, Helmholtz Munich, Neuherberg, Germany.
TUM School of Natural Sciences, Department of Bioscience, Bayerisches NMR Zentrum, Technical University of Munich, Garching, Germany.
Nat Comput Sci. 2024 May;4(5):367-378. doi: 10.1038/s43588-024-00627-2. Epub 2024 May 10.
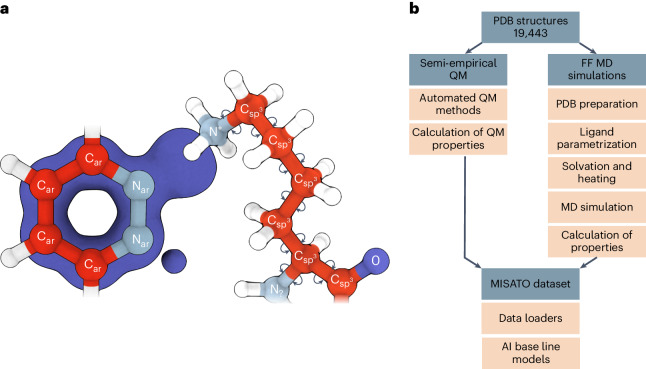
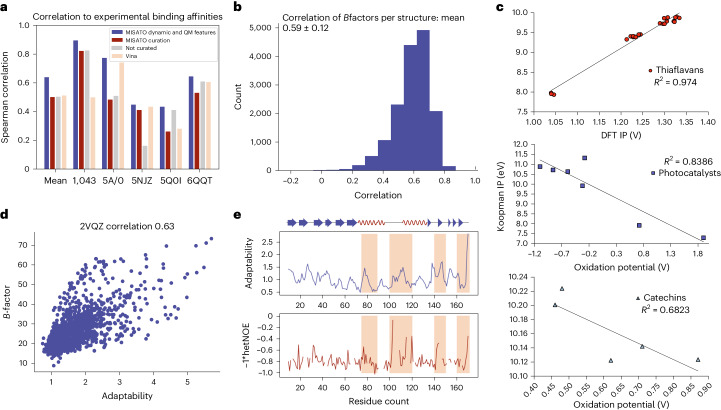
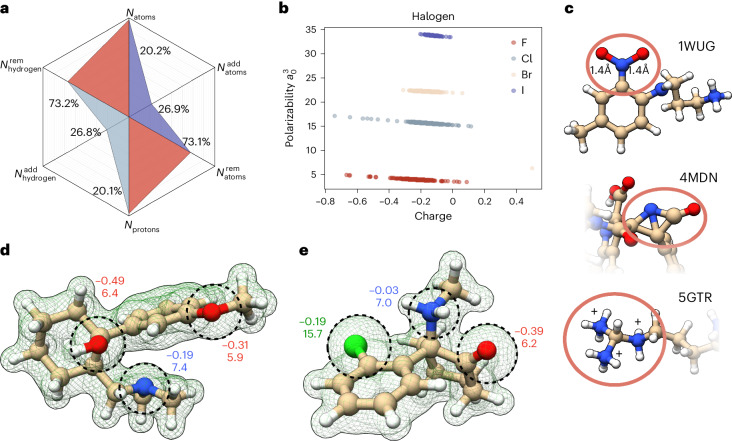
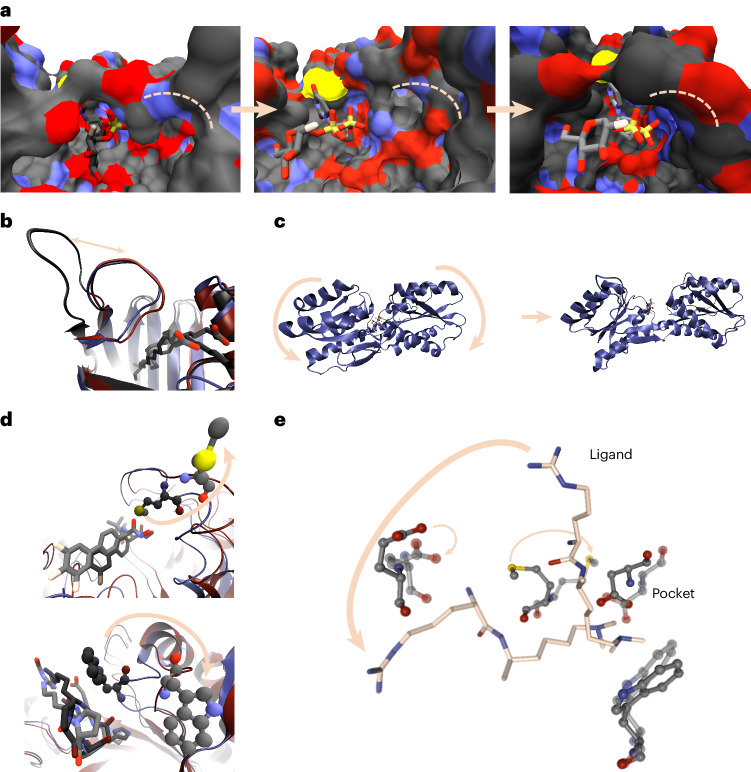
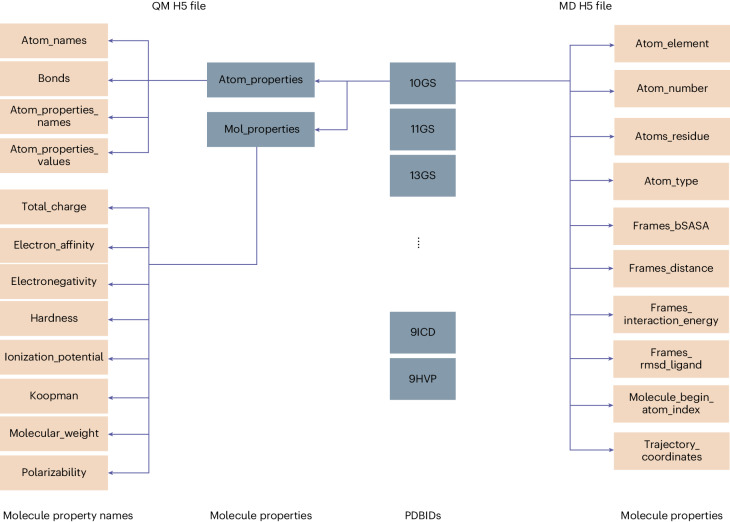
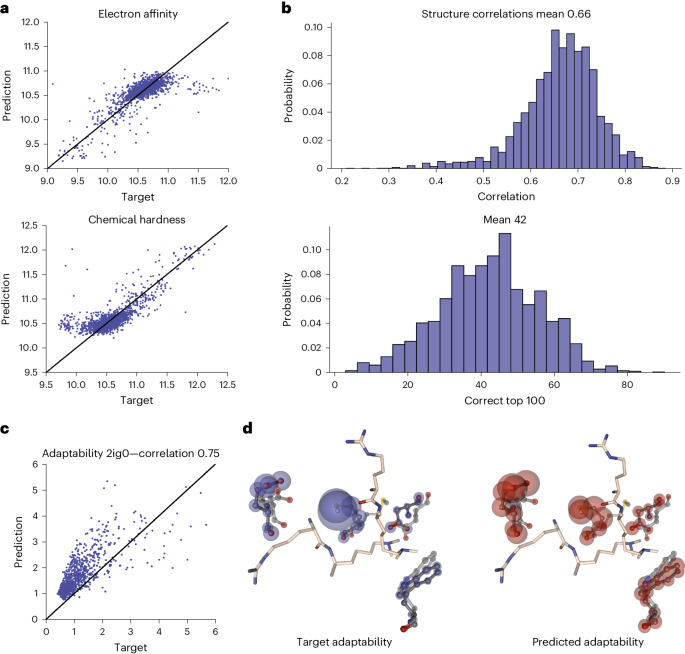
Large language models have greatly enhanced our ability to understand biology and chemistry, yet robust methods for structure-based drug discovery, quantum chemistry and structural biology are still sparse. Precise biomolecule-ligand interaction datasets are urgently needed for large language models. To address this, we present MISATO, a dataset that combines quantum mechanical properties of small molecules and associated molecular dynamics simulations of ~20,000 experimental protein-ligand complexes with extensive validation of experimental data. Starting from the existing experimental structures, semi-empirical quantum mechanics was used to systematically refine these structures. A large collection of molecular dynamics traces of protein-ligand complexes in explicit water is included, accumulating over 170 μs. We give examples of machine learning (ML) baseline models proving an improvement of accuracy by employing our data. An easy entry point for ML experts is provided to enable the next generation of drug discovery artificial intelligence models.
大型语言模型大大增强了我们对生物学和化学的理解能力,但基于结构的药物发现、量子化学和结构生物学的强大方法仍然很少。大型语言模型迫切需要精确的生物分子-配体相互作用数据集。为了解决这个问题,我们提出了 MISATO 数据集,该数据集结合了小分子的量子力学性质和~20000 个实验蛋白质-配体复合物的相关分子动力学模拟,并对实验数据进行了广泛的验证。从现有的实验结构开始,半经验量子力学被用来系统地改进这些结构。还包括一个包含超过 170μs 的蛋白质-配体复合物在明水环境中分子动力学轨迹的大型集合。我们提供了一些机器学习 (ML) 基线模型的示例,证明了通过使用我们的数据可以提高准确性。我们为 ML 专家提供了一个简单的切入点,以实现下一代药物发现人工智能模型。