Faculty of Information Technology, Beijing University of Technology, No. 100, Pingleyuan, Chaoyang District, Beijing, 100124, China.
Beijing International Collaboration Base on Brain Informatics and Wisdom Services, Beijing University of Technology, No. 100, Pingleyuan, Chaoyang District, Beijing, 100124, China.
BMC Bioinformatics. 2022 Sep 7;23(1):367. doi: 10.1186/s12859-022-04905-6.
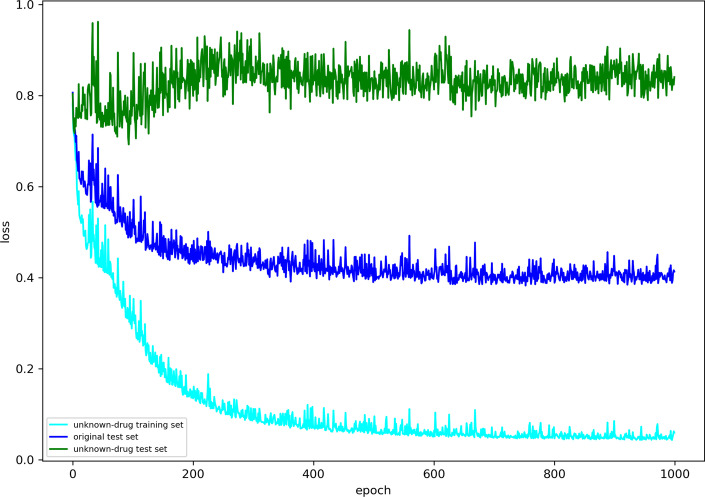
Accurately predicting drug-target binding affinity (DTA) in silico plays an important role in drug discovery. Most of the computational methods developed for predicting DTA use machine learning models, especially deep neural networks, and depend on large-scale labelled data. However, it is difficult to learn enough feature representation from tens of millions of compounds and hundreds of thousands of proteins only based on relatively limited labelled drug-target data. There are a large number of unknown drugs, which never appear in the labelled drug-target data. This is a kind of out-of-distribution problems in bio-medicine. Some recent studies adopted self-supervised pre-training tasks to learn structural information of amino acid sequences for enhancing the feature representation of proteins. However, the task gap between pre-training and DTA prediction brings the catastrophic forgetting problem, which hinders the full application of feature representation in DTA prediction and seriously affects the generalization capability of models for unknown drug discovery.
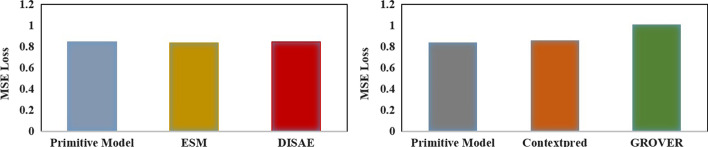
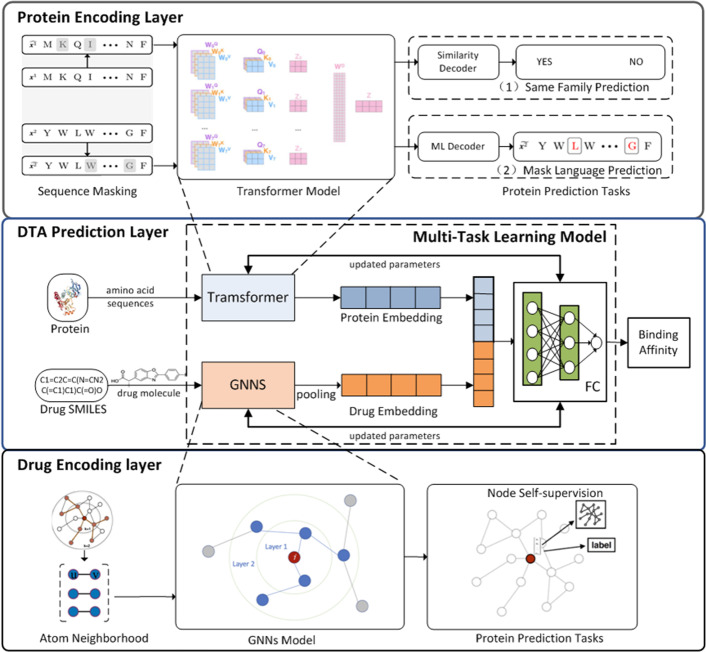
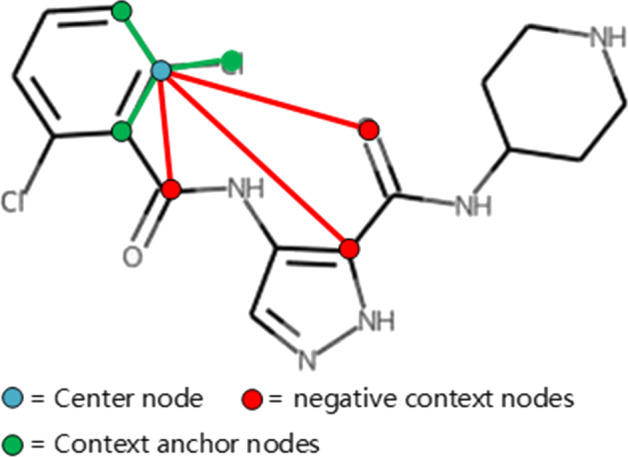
To address these problems, we propose the GeneralizedDTA, which is a new DTA prediction model oriented to unknown drug discovery, by combining pre-training and multi-task learning. We introduce self-supervised protein and drug pre-training tasks to learn richer structural information from amino acid sequences of proteins and molecular graphs of drug compounds, in order to alleviate the problem of high variance caused by encoding based on deep neural networks and accelerate the convergence of prediction model on small-scale labelled data. We also develop a multi-task learning framework with a dual adaptation mechanism to narrow the task gap between pre-training and prediction for preventing overfitting and improving the generalization capability of DTA prediction model on unknown drug discovery. To validate the effectiveness of our model, we construct an unknown drug data set to simulate the scenario of unknown drug discovery. Compared with existing DTA prediction models, the experimental results show that our model has the higher generalization capability in the DTA prediction of unknown drugs.
The advantages of our model are mainly attributed to two kinds of pre-training tasks and the multi-task learning framework, which can learn richer structural information of proteins and drugs from large-scale unlabeled data, and then effectively integrate it into the downstream prediction task for obtaining a high-quality DTA prediction in unknown drug discovery.
准确地在计算机上预测药物-靶标结合亲和力(DTA)在药物发现中起着重要作用。大多数用于预测 DTA 的计算方法都使用机器学习模型,尤其是深度神经网络,并依赖于大规模的标记数据。然而,仅基于相对有限的标记药物-靶标数据,从数千万种化合物和数十万种蛋白质中学习足够的特征表示是很困难的。有大量未知药物从未出现在标记的药物-靶标数据中。这是生物医学中一种分布外问题。一些最近的研究采用自监督预训练任务来学习氨基酸序列的结构信息,以增强蛋白质的特征表示。然而,预训练和 DTA 预测之间的任务差距带来了灾难性遗忘问题,这阻碍了特征表示在 DTA 预测中的充分应用,并严重影响了模型对未知药物发现的泛化能力。
为了解决这些问题,我们提出了一种新的面向未知药物发现的 DTA 预测模型 GeneralizedDTA,该模型通过结合预训练和多任务学习来实现。我们引入了自监督的蛋白质和药物预训练任务,从蛋白质的氨基酸序列和药物化合物的分子图中学习更丰富的结构信息,以减轻基于深度神经网络的编码引起的高方差问题,并加速预测模型在小规模标记数据上的收敛。我们还开发了一种具有双重自适应机制的多任务学习框架,以缩小预训练和预测之间的任务差距,防止过拟合,并提高 DTA 预测模型在未知药物发现中的泛化能力。为了验证我们模型的有效性,我们构建了一个未知药物数据集来模拟未知药物发现的场景。与现有的 DTA 预测模型相比,实验结果表明,我们的模型在未知药物的 DTA 预测中具有更高的泛化能力。
我们模型的优势主要归因于两种预训练任务和多任务学习框架,它们可以从大规模未标记数据中学习蛋白质和药物的更丰富的结构信息,然后将其有效地整合到下游预测任务中,从而在未知药物发现中获得高质量的 DTA 预测。