Ph.D. Program in Computer Science, The Graduate Center, The City University of New York, New York, New York 10016, United States.
Ph.D. Program in Biochemistry, The Graduate Center, The City University of New York, New York, New York 10016, United States.
J Chem Inf Model. 2021 Apr 26;61(4):1570-1582. doi: 10.1021/acs.jcim.0c01285. Epub 2021 Mar 23.
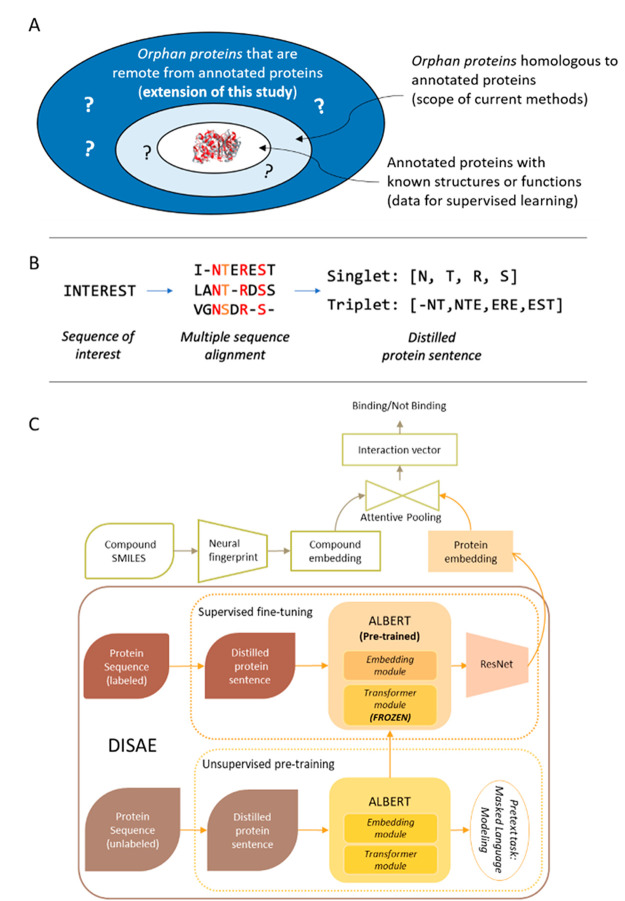
Small molecules play a critical role in modulating biological systems. Knowledge of chemical-protein interactions helps address fundamental and practical questions in biology and medicine. However, with the rapid emergence of newly sequenced genes, the endogenous or surrogate ligands of a vast number of proteins remain unknown. Homology modeling and machine learning are two major methods for assigning new ligands to a protein but mostly fail when sequence homology between an unannotated protein and those with known functions or structures is low. In this study, we develop a new deep learning framework to predict chemical binding to evolutionary divergent unannotated proteins, whose ligand cannot be reliably predicted by existing methods. By incorporating evolutionary information into self-supervised learning of unlabeled protein sequences, we develop a novel method, distilled sequence alignment embedding (DISAE), for the protein sequence representation. DISAE can utilize all protein sequences and their multiple sequence alignment (MSA) to capture functional relationships between proteins without the knowledge of their structure and function. Followed by the DISAE pretraining, we devise a module-based fine-tuning strategy for the supervised learning of chemical-protein interactions. In the benchmark studies, DISAE significantly improves the generalizability of machine learning models and outperforms the state-of-the-art methods by a large margin. Comprehensive ablation studies suggest that the use of MSA, sequence distillation, and triplet pretraining critically contributes to the success of DISAE. The interpretability analysis of DISAE suggests that it learns biologically meaningful information. We further use DISAE to assign ligands to human orphan G-protein coupled receptors (GPCRs) and to cluster the human GPCRome by integrating their phylogenetic and ligand relationships. The promising results of DISAE open an avenue for exploring the chemical landscape of entire sequenced genomes.
小分子在调节生物系统中起着至关重要的作用。对化学-蛋白质相互作用的了解有助于解决生物学和医学中的基础和实际问题。然而,随着新测序基因的迅速出现,大量蛋白质的内源性或替代配体仍然未知。同源建模和机器学习是为蛋白质分配新配体的两种主要方法,但当未注释蛋白质与具有已知功能或结构的蛋白质之间的序列同源性较低时,这两种方法大多会失败。在这项研究中,我们开发了一种新的深度学习框架,用于预测化学结合进化上不同的未注释蛋白质,对于这些蛋白质,现有方法无法可靠地预测其配体。通过将进化信息纳入未标记蛋白质序列的自监督学习中,我们开发了一种新的方法,即蒸馏序列对齐嵌入(Distilled Sequence Alignment Embedding,DISAE),用于蛋白质序列表示。DISAE 可以利用所有蛋白质序列及其多重序列比对(Multiple Sequence Alignment,MSA)来捕获蛋白质之间的功能关系,而无需了解其结构和功能。在 DISAE 预训练之后,我们设计了一种基于模块的微调策略,用于化学-蛋白质相互作用的监督学习。在基准研究中,DISAE 显著提高了机器学习模型的泛化能力,并以较大的优势超过了最先进的方法。全面的消融研究表明,使用 MSA、序列蒸馏和三元组预训练对 DISAE 的成功至关重要。DISAE 的可解释性分析表明,它学习了有生物学意义的信息。我们进一步使用 DISAE 为人类孤儿 G 蛋白偶联受体(GPCR)分配配体,并通过整合它们的进化和亲缘关系对人类 GPCR 组进行聚类。DISAE 的有前途的结果为探索整个测序基因组的化学景观开辟了一条途径。