Laboratory for Computational Biology & Biomolecular Design, School of Biochemical Engineering, Indian Institute of Technology (BHU), Varanasi, 221005, Uttar Pradesh, India.
Molecular and Structural Biophysics Laboratory, Department of Biochemistry, North-Eastern Hill University, Shillong, 793022, India; Regional Director's Office, Indira Gandhi National Open University, Regional Centre Kohima, Kenuozou, Kohima, 797001, India.
Biochem Biophys Res Commun. 2022 Nov 12;629:54-60. doi: 10.1016/j.bbrc.2022.09.010. Epub 2022 Sep 7.
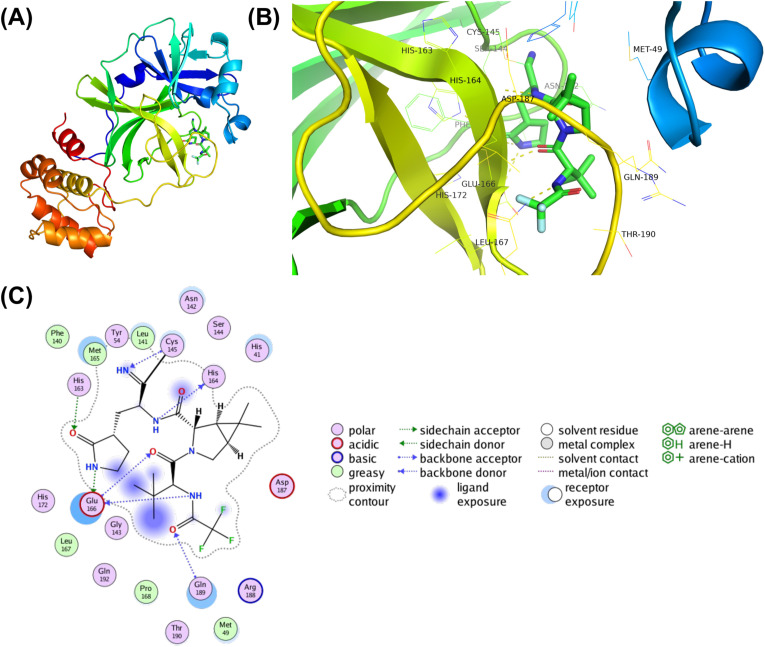
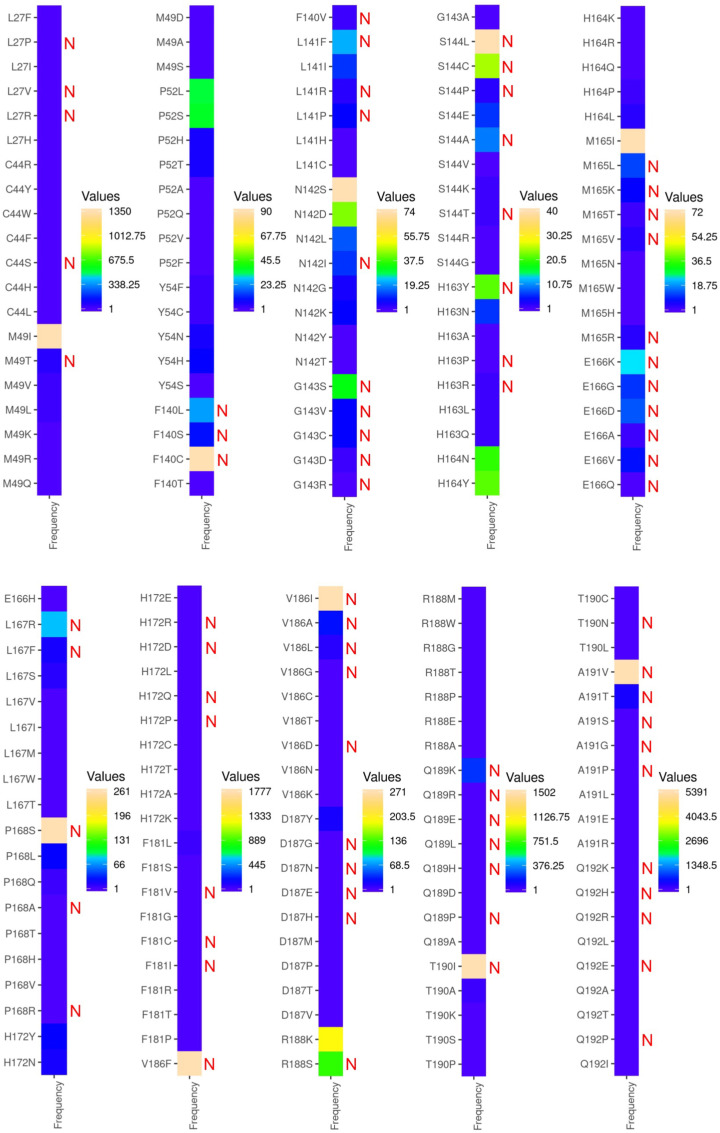
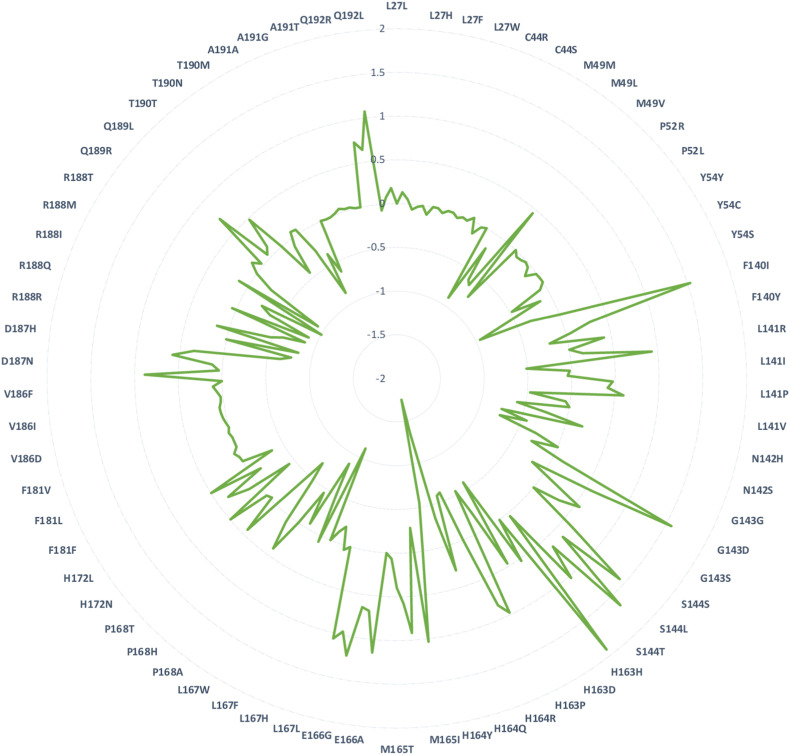
Shortly after the onset of the COVID-19 pandemic, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has acquired numerous variations in its intracellular proteins to adapt quickly, become more infectious, and ultimately develop drug resistance by mutating certain hotspot residues. To keep the emerging variants at bay, including Omicron and subvariants, FDA has approved the antiviral nirmatrelvir for mild-to-moderate and high-risk COVID-19 cases. Like other viruses, SARS-CoV-2 could acquire mutations in its main protease (M) to adapt and develop resistance against nirmatrelvir. Employing a unique high-throughput protein design technique, the hotspot residues, and signatures of adaptation of M having the highest probability of mutating and rendering nirmatrelvir ineffective were identified. Our results show that ∼40% of the designed mutations in M already exist in the globally circulating SARS-CoV-2 lineages and several predicted mutations. Moreover, several high-frequency, designed mutations were found to be in corroboration with the experimentally reported nirmatrelvir-resistant mutants and are naturally occurring. Our work on the targeted design of the nirmatrelvir-binding site offers a comprehensive picture of potential hotspot sites and resistance mutations in M and is thus crucial in comprehending viral adaptation, robust antiviral design, and surveillance of evolving M variations.
在 COVID-19 大流行开始后不久,严重急性呼吸综合征冠状病毒 2(SARS-CoV-2)在其细胞内蛋白中获得了许多变异,以快速适应、变得更具传染性,并通过突变某些热点残基最终产生耐药性。为了控制包括奥密克戎和亚变种在内的新出现的变种,FDA 已批准抗病毒药物奈玛特韦用于轻度至中度和高危 COVID-19 病例。与其他病毒一样,SARS-CoV-2 可能会在其主要蛋白酶(M)中发生突变,以适应并对奈玛特韦产生耐药性。采用独特的高通量蛋白质设计技术,确定了 M 中具有最高突变可能性和使奈玛特韦失效的热点残基和适应特征。我们的研究结果表明,全球循环的 SARS-CoV-2 谱系和几种预测的突变中已经存在 M 中约 40%的设计突变。此外,还发现了几个高频设计突变与实验报告的奈玛特韦耐药突变体相吻合,并且是自然发生的。我们对奈玛特韦结合位点的靶向设计工作提供了 M 中潜在热点位点和耐药突变的全面图景,因此对于理解病毒适应、强大的抗病毒设计和不断进化的 M 变异监测至关重要。