Division of Genome Analysis Platform Development, National Cancer Center Research Institute, Tokyo, Japan.
Cancer RNA Research Unit, National Cancer Center Research Institute, Tokyo, Japan.
Nat Commun. 2022 Sep 29;13(1):5357. doi: 10.1038/s41467-022-32887-9.
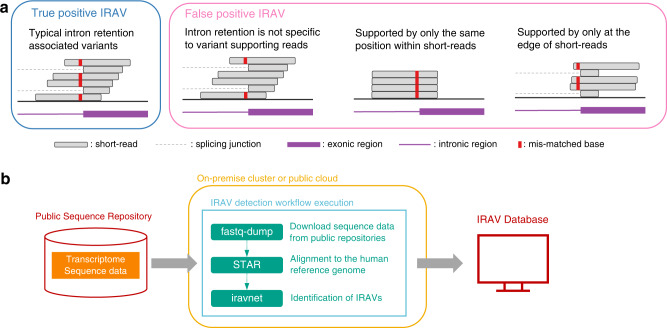
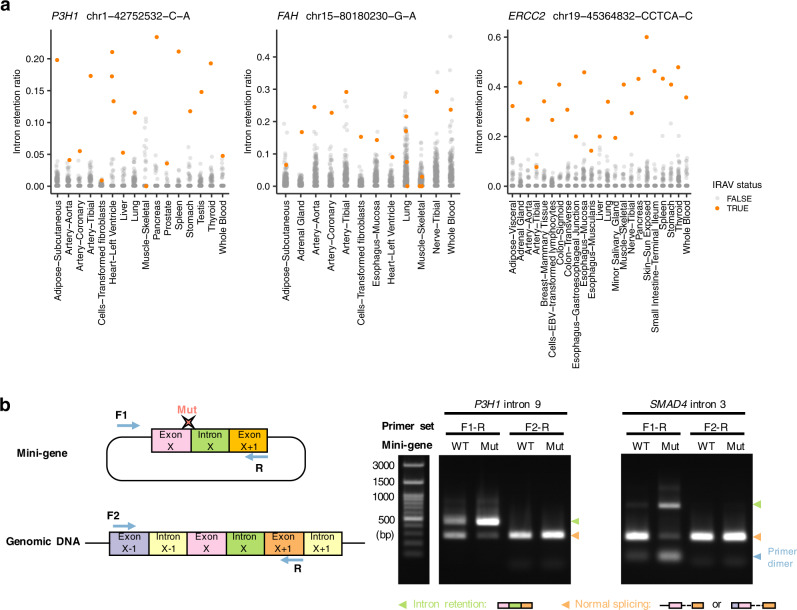
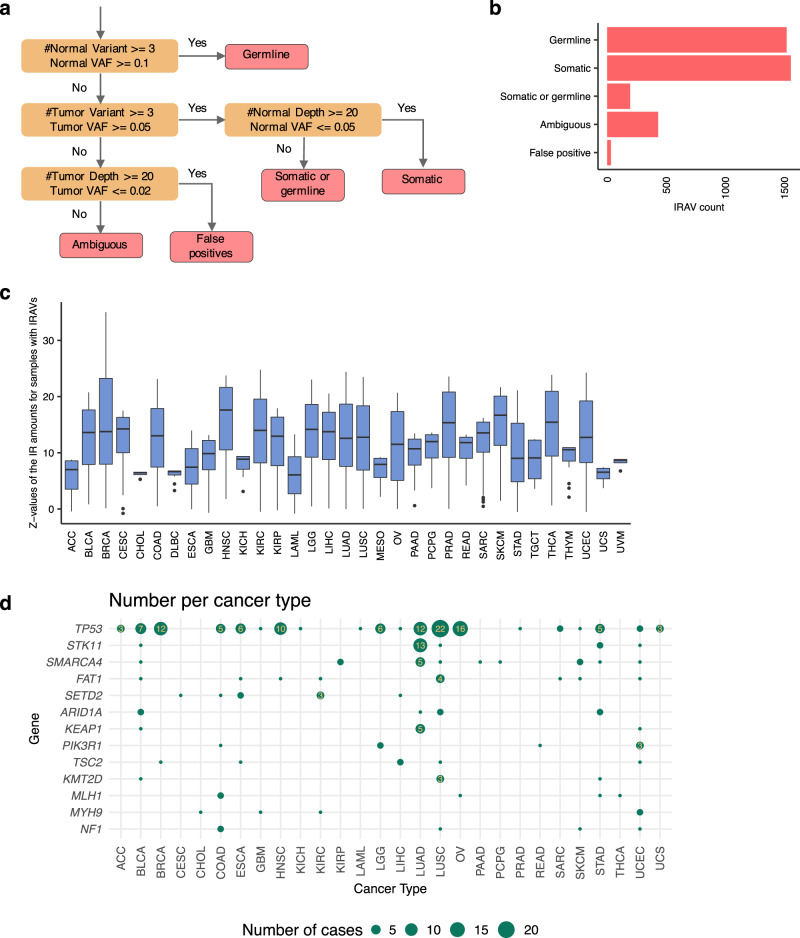
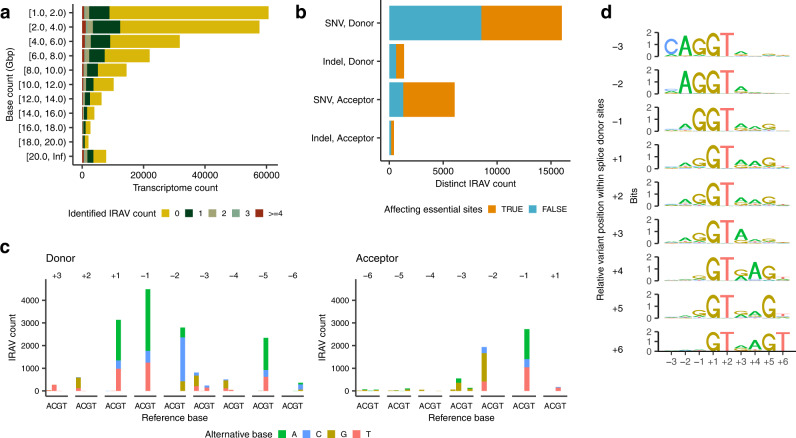
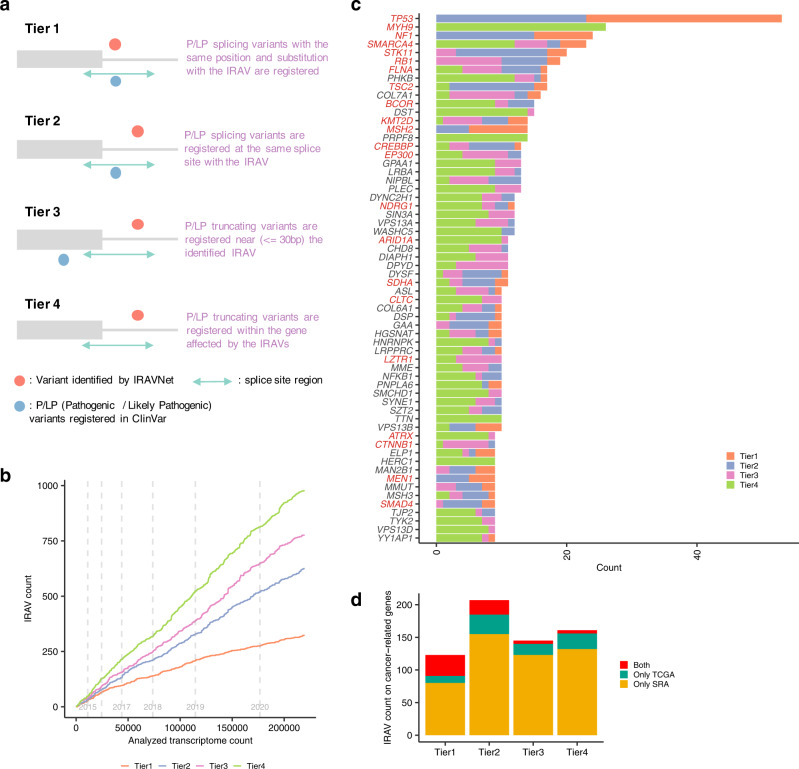
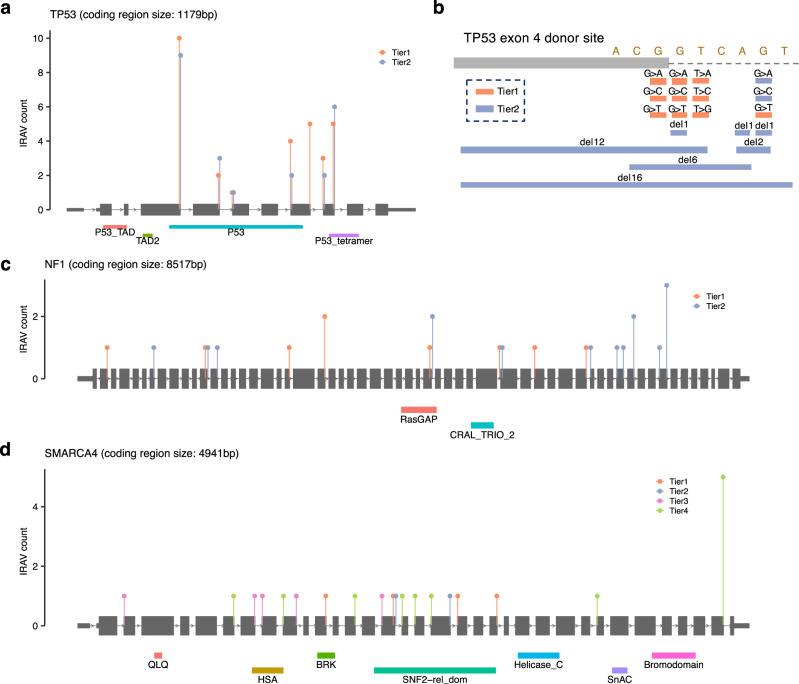
Many disease-associated genomic variants disrupt gene function through abnormal splicing. With the advancement of genomic medicine, identifying disease-associated splicing associated variants has become more important than ever. Most bioinformatics approaches to detect splicing associated variants require both genome and transcriptomic data. However, there are not many datasets where both of them are available. In this study, we develop a methodology to detect genomic variants that cause splicing changes (more specifically, intron retention), using transcriptome sequencing data alone. After evaluating its sensitivity and precision, we apply it to 230,988 transcriptome sequencing data from the publicly available repository and identified 27,049 intron retention associated variants (IRAVs). In addition, by exploring positional relationships with variants registered in existing disease databases, we extract 3,000 putative disease-associated IRAVs, which range from cancer drivers to variants linked with autosomal recessive disorders. The in-silico screening framework demonstrates the possibility of near-automatically acquiring medical knowledge, making the most of massively accumulated publicly available sequencing data. Collections of IRAVs identified in this study are available through IRAVDB ( https://iravdb.io/ ).
许多与疾病相关的基因组变异通过异常剪接破坏基因功能。随着基因组医学的进步,鉴定与剪接相关的疾病相关变异变得比以往任何时候都更加重要。大多数用于检测剪接相关变异的生物信息学方法都需要基因组和转录组数据。然而,有很多数据集同时提供这两种数据。在这项研究中,我们开发了一种仅使用转录组测序数据检测导致剪接变化(更具体地说是内含子保留)的基因组变异的方法。在评估其灵敏度和精度后,我们将其应用于来自公开存储库的 230988 个转录组测序数据,并鉴定出 27049 个内含子保留相关变异(IRAV)。此外,通过探索与现有疾病数据库中注册的变异的位置关系,我们提取了 3000 个潜在的与疾病相关的 IRAV,其中包括癌症驱动因子和与常染色体隐性疾病相关的变异。该计算筛选框架展示了充分利用大量积累的公开可用测序数据自动获取医学知识的可能性。本研究中鉴定的 IRAV 集合可通过 IRAVDB(https://iravdb.io/)获得。