Systems Biology of Infection Laboratory, Department of Biochemistry and Molecular Biology, Biosciences Faculty, Universitat Autònoma de Barcelona, 08193 Bellaterra, Spain.
Int J Mol Sci. 2022 Sep 29;23(19):11489. doi: 10.3390/ijms231911489.
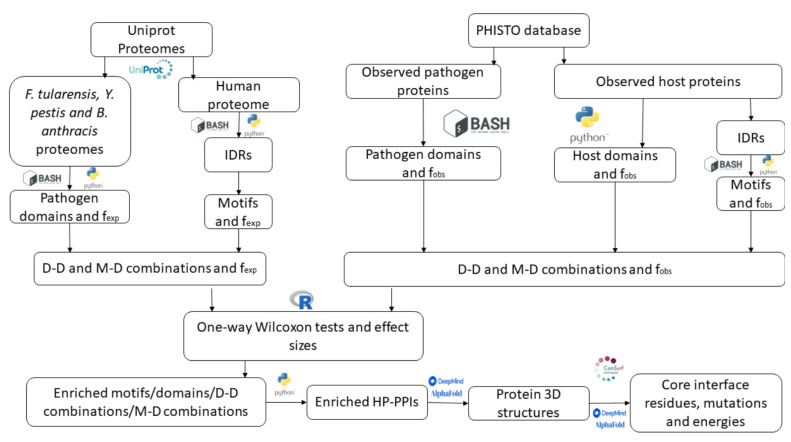
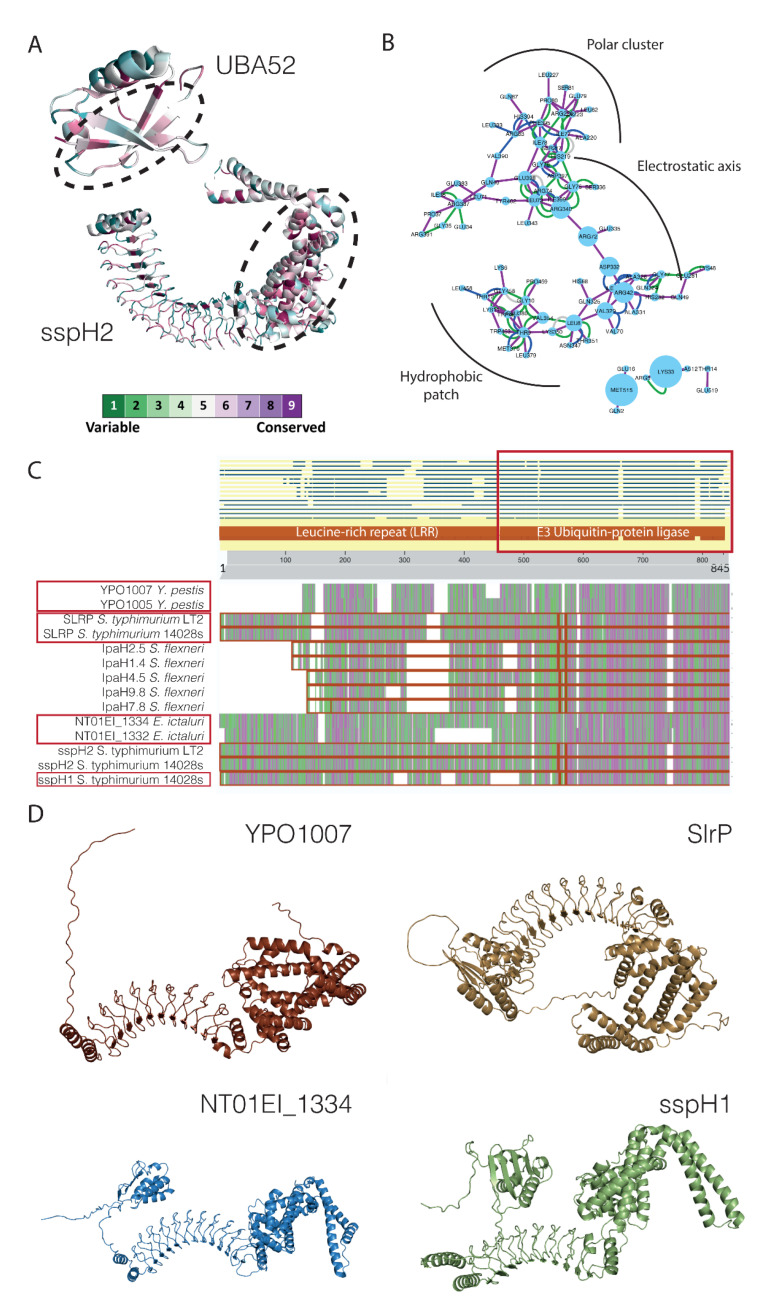
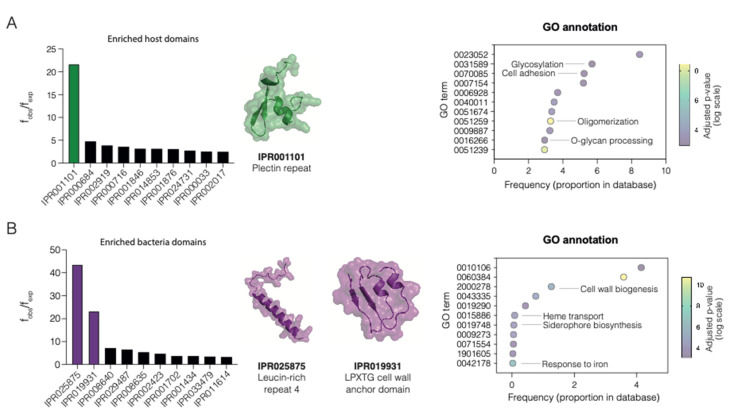
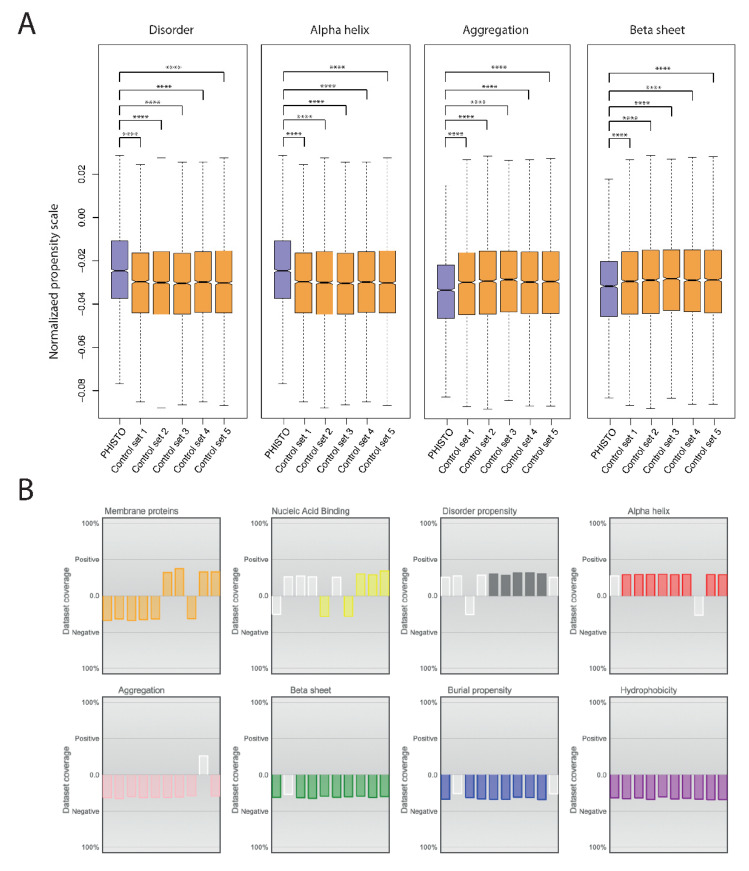
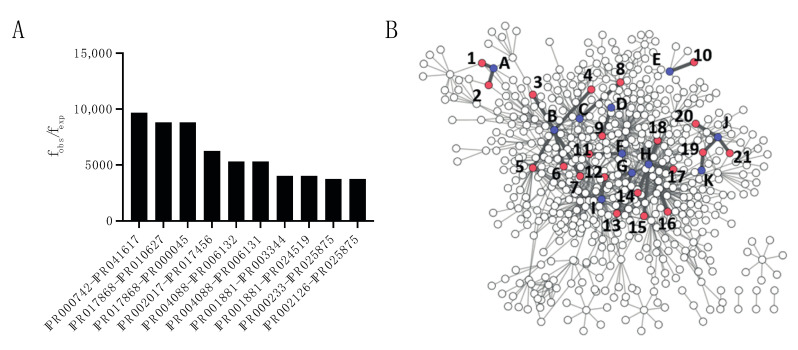
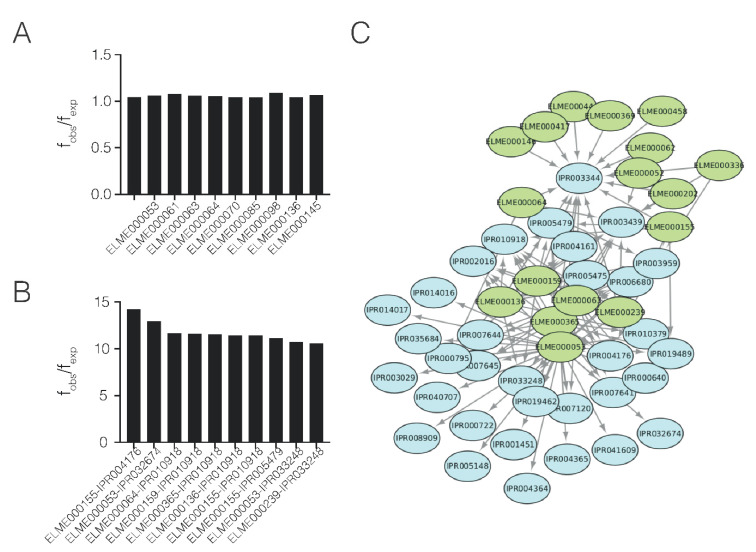
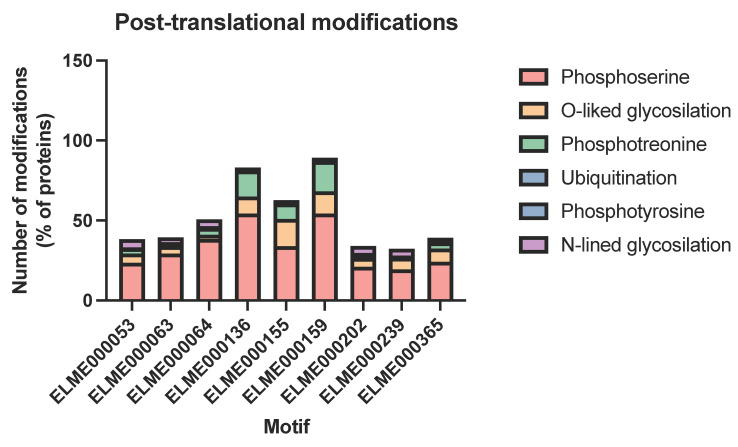
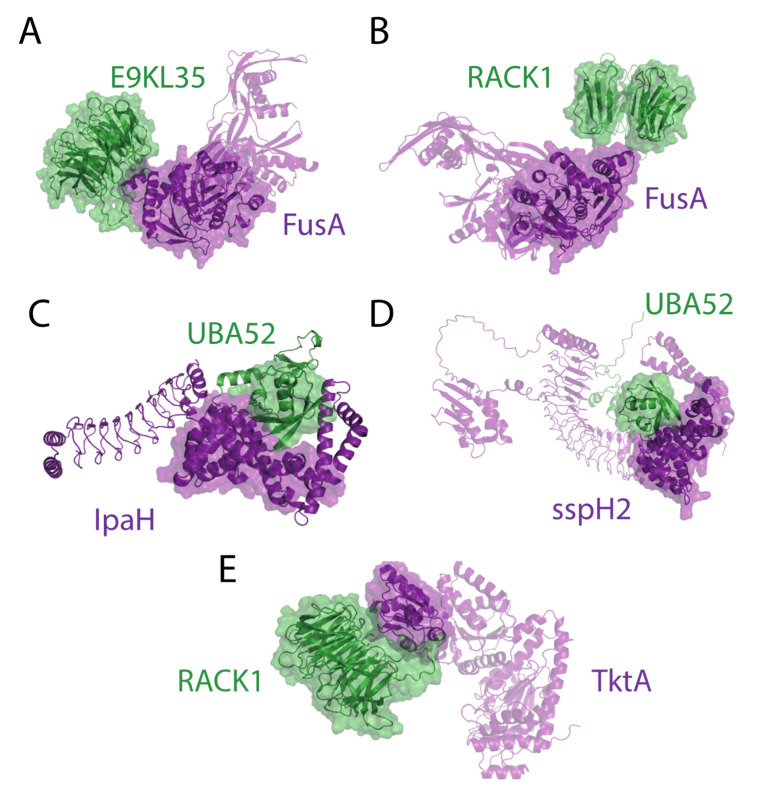
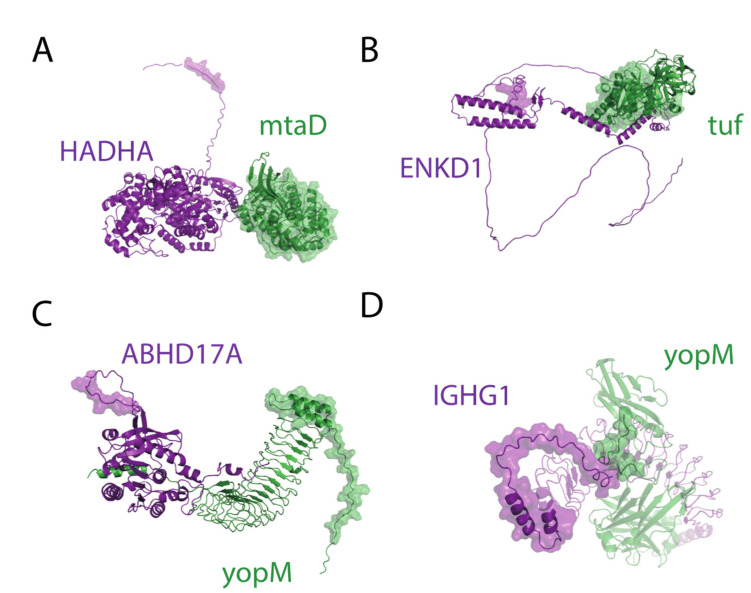
Adhesion and colonization of host cells by pathogenic bacteria depend on protein-protein interactions (PPIs). These interactions are interesting from the pharmacological point of view since new molecules that inhibit host-pathogen PPIs would act as new antimicrobials. Most of these interactions are discovered using high-throughput methods that may display a high false positive rate. The absence of curation of these databases can make the available data unreliable. To address this issue, a comprehensive filtering process was developed to obtain a reliable list of domains and motifs that participate in PPIs between bacteria and human cells. From a structural point of view, our analysis revealed that human proteins involved in the interactions are rich in alpha helix and disordered regions and poorer in beta structure. Disordered regions in human proteins harbor short sequence motifs that are specifically recognized by certain domains in pathogenic proteins. The most relevant domain-domain interactions were validated by AlphaFold, showing that a proper analysis of host-pathogen PPI databases can reveal structural conserved patterns. Domain-motif interactions, on the contrary, were more difficult to validate, since unstructured regions were involved, where AlphaFold could not make a good prediction. Moreover, these interactions are also likely accommodated by post-translational modifications, especially phosphorylation, which can potentially occur in 25-50% of host proteins. Hence, while common structural patterns are involved in host-pathogen PPIs and can be retrieved from available databases, more information is required to properly infer the full interactome. By resolving these issues, and in combination with new prediction tools like Alphafold, new classes of antimicrobials could be discovered from a more detailed understanding of these interactions.
宿主细胞与病原菌的黏附和定植依赖于蛋白-蛋白相互作用(PPIs)。从药理学角度来看,这些相互作用很有趣,因为抑制宿主-病原体 PPI 的新分子将作为新的抗菌药物。这些相互作用中的大多数是使用高通量方法发现的,这些方法可能显示出高的假阳性率。这些数据库缺乏整理,会使得可用数据变得不可靠。为了解决这个问题,开发了一个全面的过滤过程,以获得参与细菌和人类细胞之间 PPI 的可靠结构域和基序列表。从结构的角度来看,我们的分析表明,参与相互作用的人类蛋白富含α螺旋和无规则区域,β结构较少。人类蛋白中的无规则区域含有短序列基序,这些基序被病原体蛋白中的某些结构域特异性识别。通过 AlphaFold 验证了最相关的结构域-结构域相互作用,表明对宿主-病原体 PPI 数据库进行适当的分析可以揭示结构保守模式。相反,结构域-基序相互作用更难验证,因为涉及无规则区域,而 AlphaFold 无法对此做出很好的预测。此外,这些相互作用也可能被翻译后修饰(如磷酸化)所适应,在 25-50%的宿主蛋白中可能发生磷酸化。因此,虽然宿主-病原体 PPI 涉及常见的结构模式,可以从现有数据库中检索到,但为了正确推断全互作网络,还需要更多的信息。通过解决这些问题,并结合像 AlphaFold 这样的新预测工具,可以从对这些相互作用的更详细了解中发现新的抗菌药物类别。