Bettink Stephanie I, Reil Jan-Christian, Kazakov Andrey, Körbel Christina, Millenaar Dominic, Laufs Ulrich, Scheller Bruno, Böhm Michael, Schirmer Stephan H
Internal Medicine III, Saarland University, 66421 Homburg, Germany.
Medical Clinic II, University Hospital Schleswig-Holstein, 23538 Lübeck, Germany.
Biomedicines. 2022 Oct 19;10(10):2636. doi: 10.3390/biomedicines10102636.
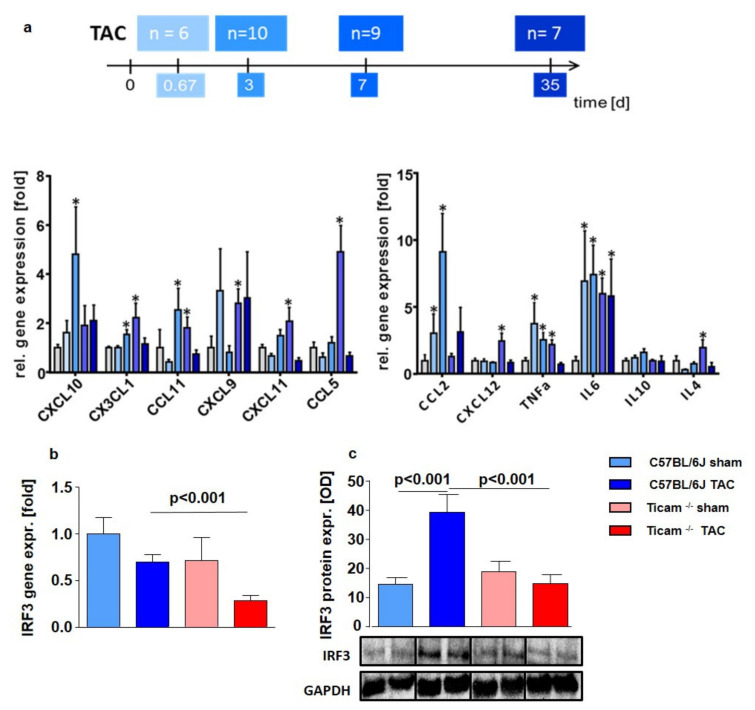
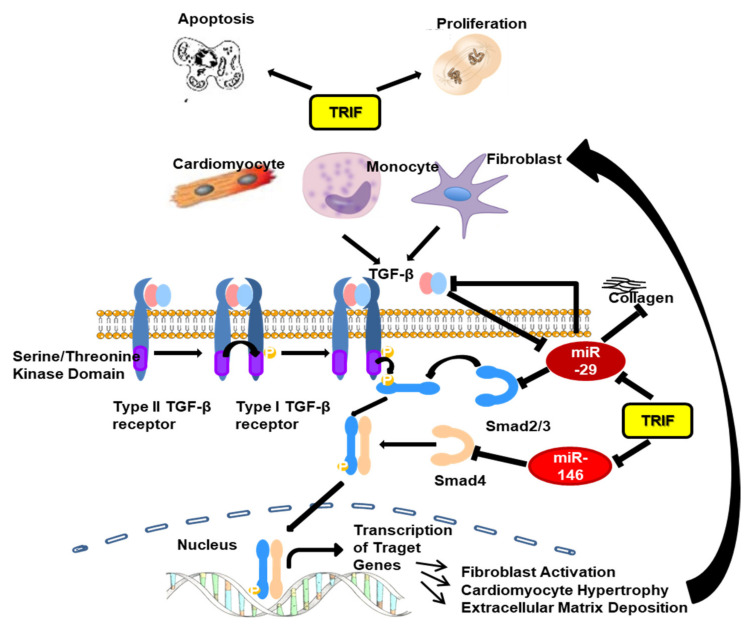
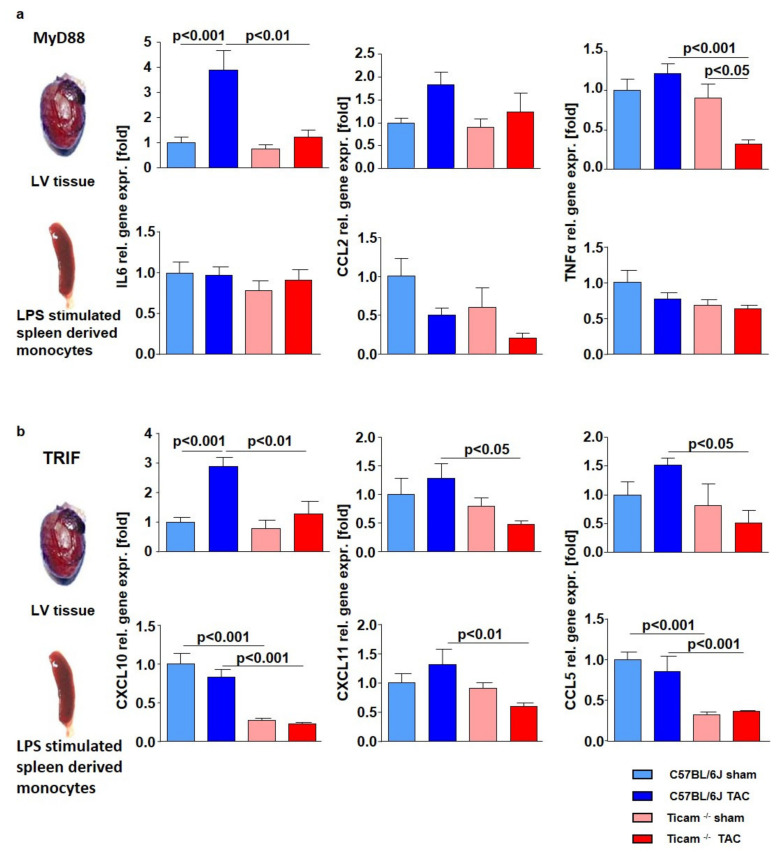
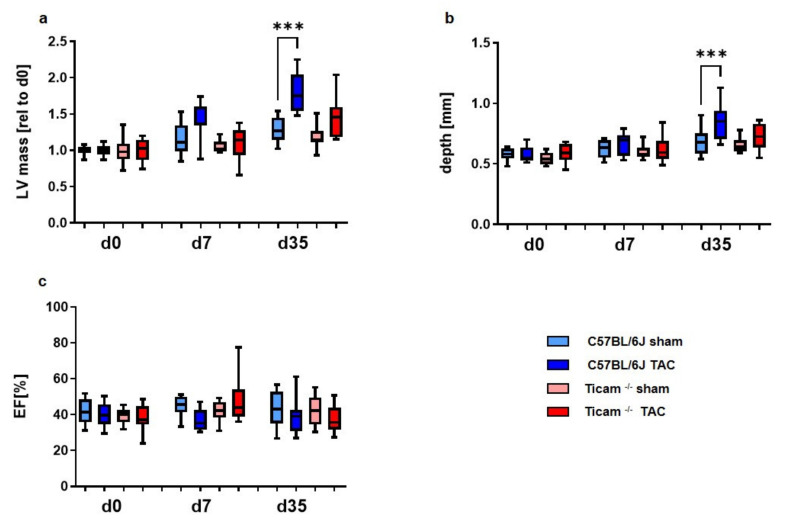
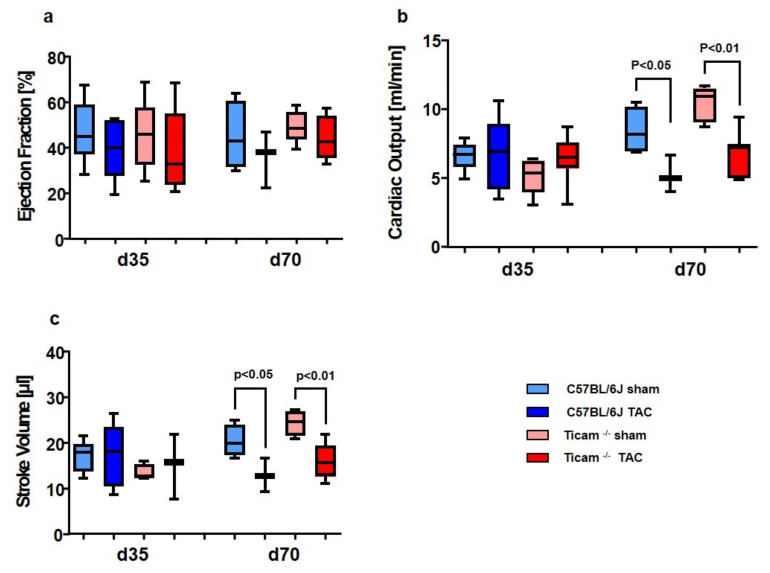
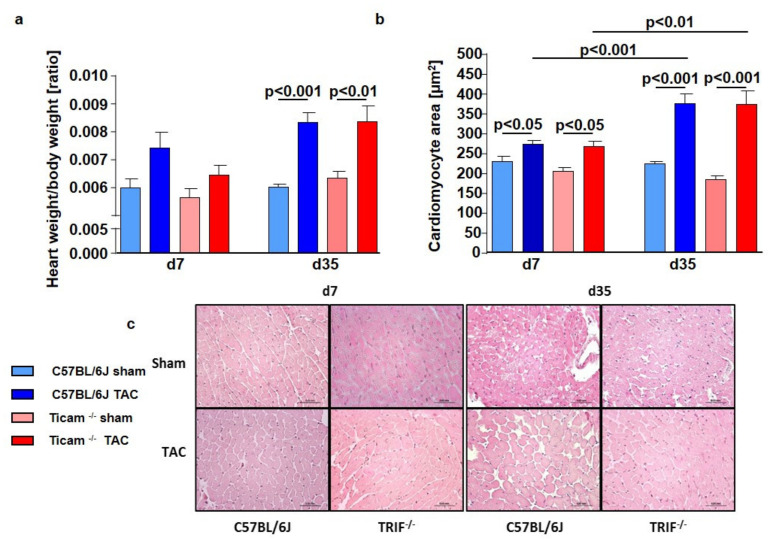
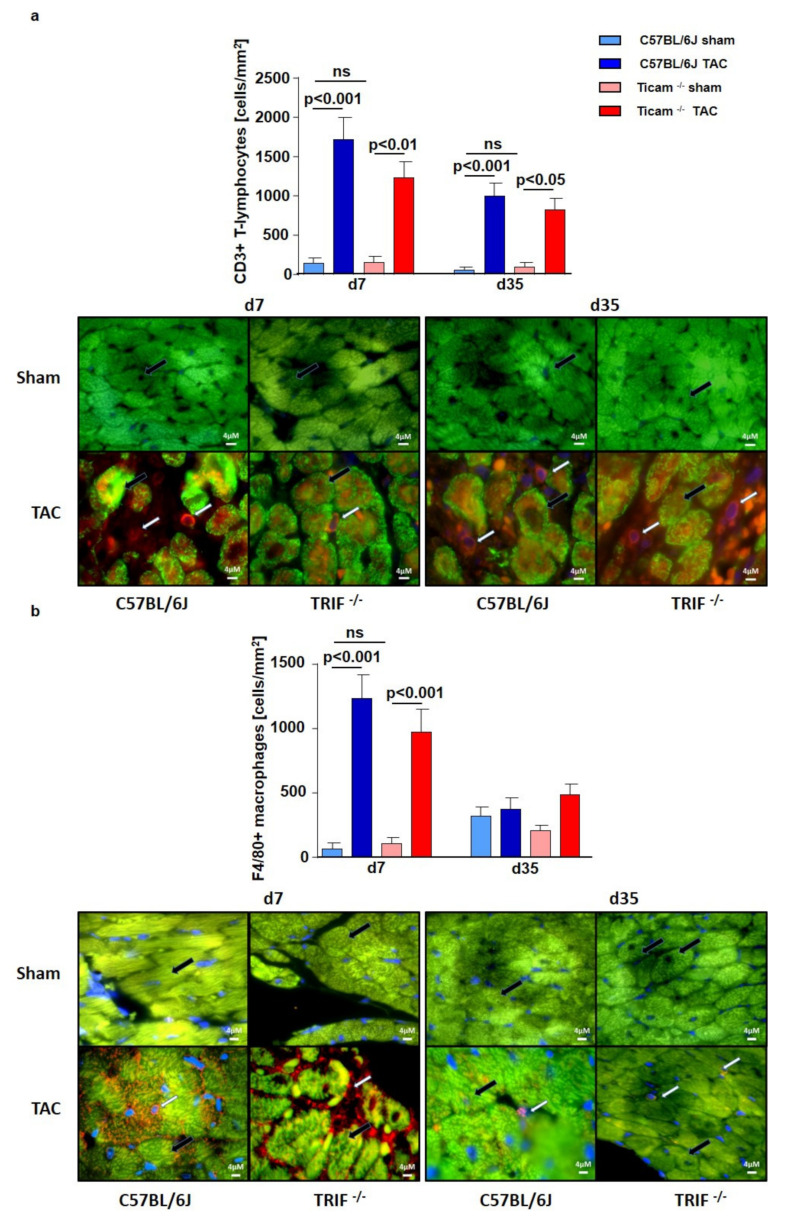
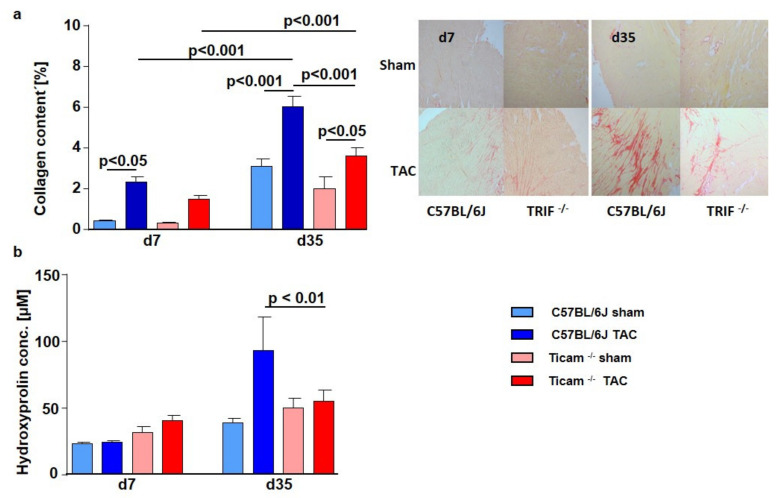
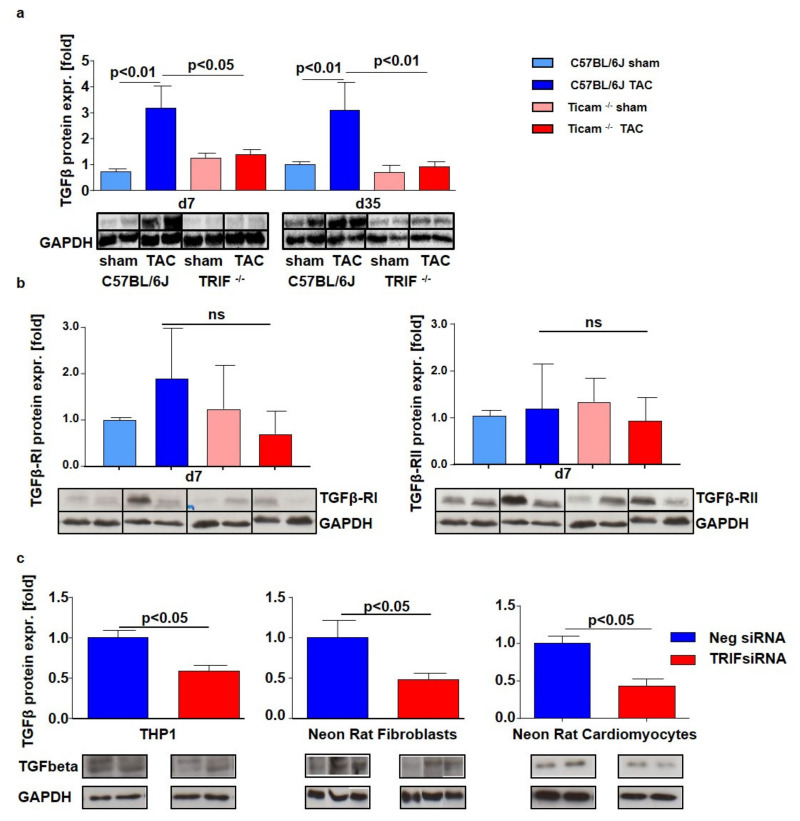
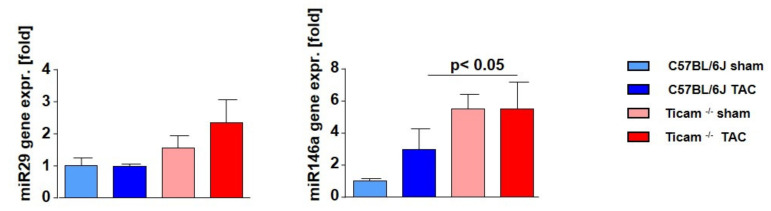
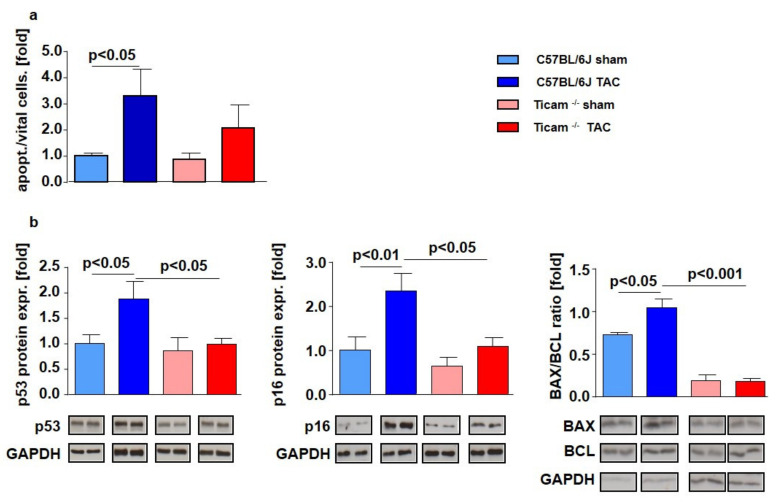
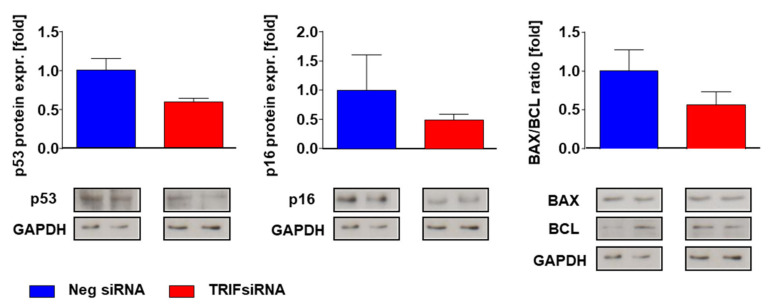
Pressure-overload-induced cardiac hypertrophy represents one cause of the development of heart failure. The aim of this study is to characterize the influence of the TIR-domain-containing adapter-inducing interferon-β (TRIF) during afterload-induced myocardial remodeling. After trans-aortic constriction (TAC), cardiac pressure overload leads to an early increase in MyD88- (Myeloid differentiation primary response gene 88) and TRIF-dependent cytokines. The maximum cytokine expression appeared within the first week and decreased to its control level within five weeks. While cardiomyocyte hypertrophy was comparable, the myocardial accumulation of the inflammatory cells was lower in TRIFmice. At d7, TRIF deficiency reduced transcription factors and TRIF-dependent cytokines. Through the modulation of the TGF-β-signaling pathway and anti-fibrotic microRNAs, TRIF was involved in the development of interstitial fibrosis. The absence of TRIF was associated with a decreased expression of proapoptotic proteins. In echocardiography and working heart analyses, TRIF deficiency slowed left-ventricular wall thickening, myocardial hypertrophy, and reduces the ejection fraction. In summary, TRIF is an important adapter protein for the release of inflammatory cytokines and the accumulation of inflammatory cells in the early stage of maladaptive cardiac remodeling. TRIF is involved in the development of cardiac fibrosis by modulating inflammatory and fibrotic signal transduction pathways.
压力超负荷诱导的心肌肥大是心力衰竭发展的一个原因。本研究的目的是描述含TIR结构域的衔接蛋白诱导干扰素-β(TRIF)在负荷后心肌重塑过程中的影响。经主动脉缩窄(TAC)后,心脏压力超负荷导致髓样分化初级反应基因88(MyD88)和TRIF依赖性细胞因子早期增加。细胞因子表达在第一周达到峰值,并在五周内降至对照水平。虽然心肌细胞肥大程度相当,但TRIF基因敲除小鼠的炎症细胞心肌积聚较少。在第7天,TRIF缺陷减少了转录因子和TRIF依赖性细胞因子。通过调节转化生长因子-β信号通路和抗纤维化微小RNA,TRIF参与了间质纤维化的发展。TRIF的缺失与促凋亡蛋白表达降低有关。在超声心动图和工作心脏分析中,TRIF缺陷减缓了左心室壁增厚、心肌肥大,并降低了射血分数。总之,TRIF是适应性不良心脏重塑早期炎症细胞因子释放和炎症细胞积聚的重要衔接蛋白。TRIF通过调节炎症和纤维化信号转导通路参与心脏纤维化的发展。