Wu Tian, Liu Zipeng, Mak Timothy Shin Heng, Sham Pak Chung
Department of Psychiatry, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Pok Fu Lam, Hong Kong SAR, China.
State Key Laboratory of Brain and Cognitive Sciences, The University of Hong Kong, Pok Fu Lam, Hong Kong SAR, China.
Front Genet. 2022 Oct 10;13:989639. doi: 10.3389/fgene.2022.989639. eCollection 2022.
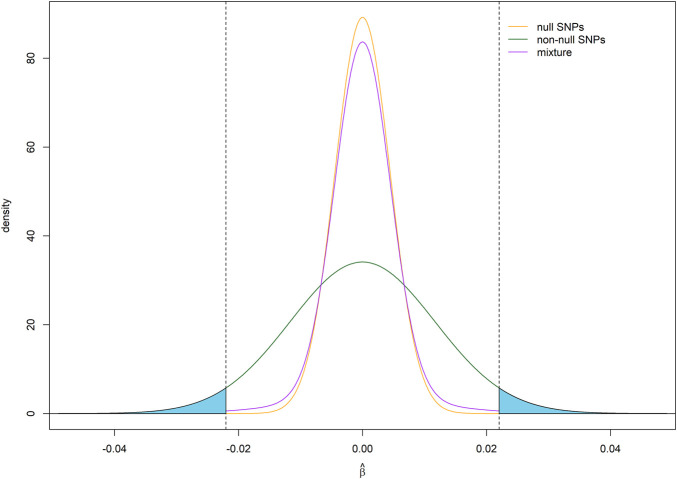
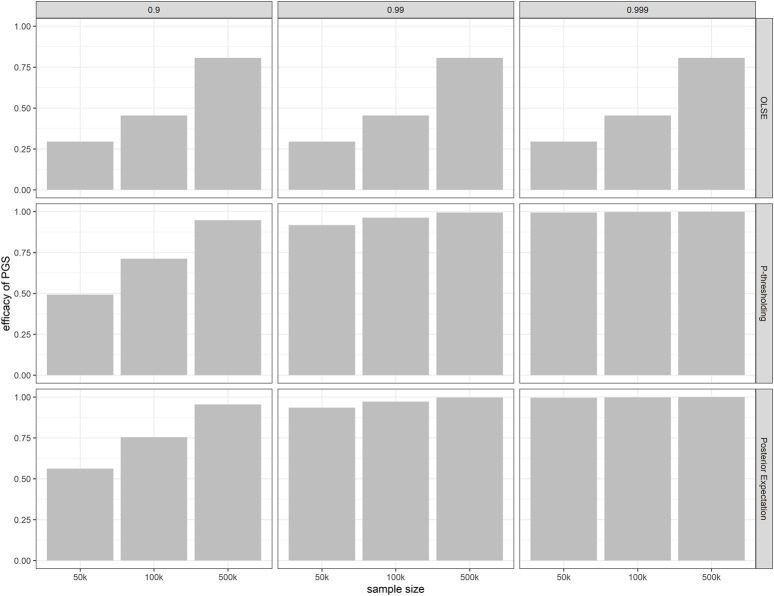
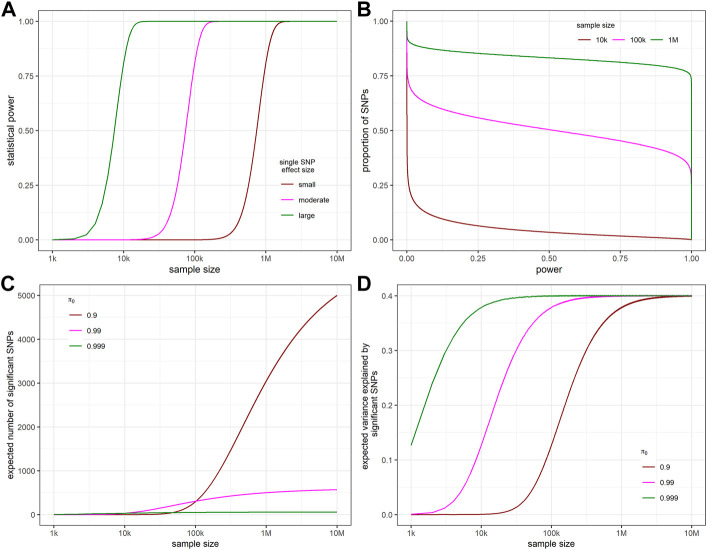
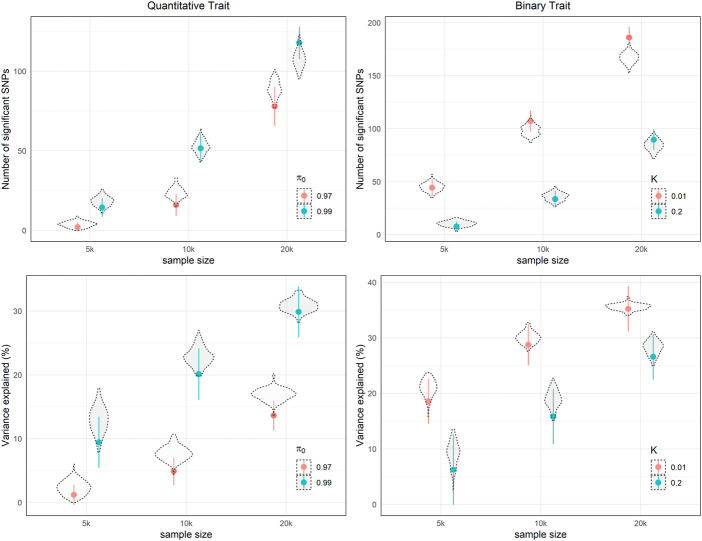
Power calculation is a necessary step when planning genome-wide association studies (GWAS) to ensure meaningful findings. Statistical power of GWAS depends on the genetic architecture of phenotype, sample size, and study design. While several computer programs have been developed to perform power calculation for single SNP association testing, it might be more appropriate for GWAS power calculation to address the probability of detecting any number of associated SNPs. In this paper, we derive the statistical power distribution across causal SNPs under the assumption of a point-normal effect size distribution. We demonstrate how key outcome indices of GWAS are related to the genetic architecture (heritability and polygenicity) of the phenotype through the power distribution. We also provide a fast, flexible and interactive power calculation tool which generates predictions for key GWAS outcomes including the number of independent significant SNPs, the phenotypic variance explained by these SNPs, and the predictive accuracy of resulting polygenic scores. These results could also be used to explore the future behaviour of GWAS as sample sizes increase further. Moreover, we present results from simulation studies to validate our derivation and evaluate the agreement between our predictions and reported GWAS results.
在规划全基因组关联研究(GWAS)时,功效计算是确保获得有意义结果的必要步骤。GWAS的统计功效取决于表型的遗传结构、样本量和研究设计。虽然已经开发了几个计算机程序来进行单核苷酸多态性(SNP)关联测试的功效计算,但对于GWAS功效计算而言,处理检测任意数量相关SNP的概率可能更为合适。在本文中,我们在点正态效应大小分布的假设下,推导出因果SNP的统计功效分布。我们展示了GWAS的关键结果指标如何通过功效分布与表型的遗传结构(遗传力和多基因性)相关联。我们还提供了一个快速、灵活且交互式的功效计算工具,该工具可为关键的GWAS结果生成预测,包括独立显著SNP的数量、这些SNP解释的表型方差以及所得多基因分数的预测准确性。这些结果还可用于探索随着样本量进一步增加GWAS未来的表现。此外,我们展示了模拟研究的结果,以验证我们的推导并评估我们的预测与报告的GWAS结果之间的一致性。