Interdisciplinary Research Center HALOmem, Charles Tanford Protein Center, Martin Luther University Halle-Wittenberg, Halle/Saale, Germany.
Institute of Biochemistry and Biotechnology, Martin Luther University Halle-Wittenberg, Halle/Saale, Germany.
Protein Sci. 2023 Jan;32(1):e4523. doi: 10.1002/pro.4523.
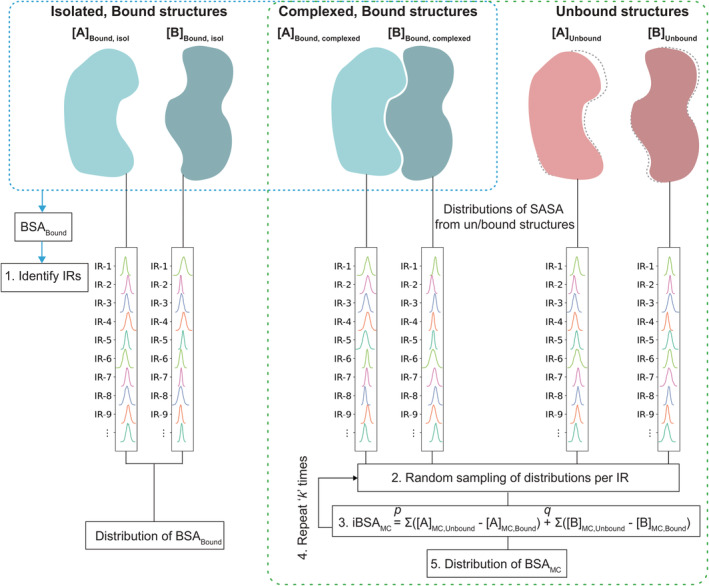
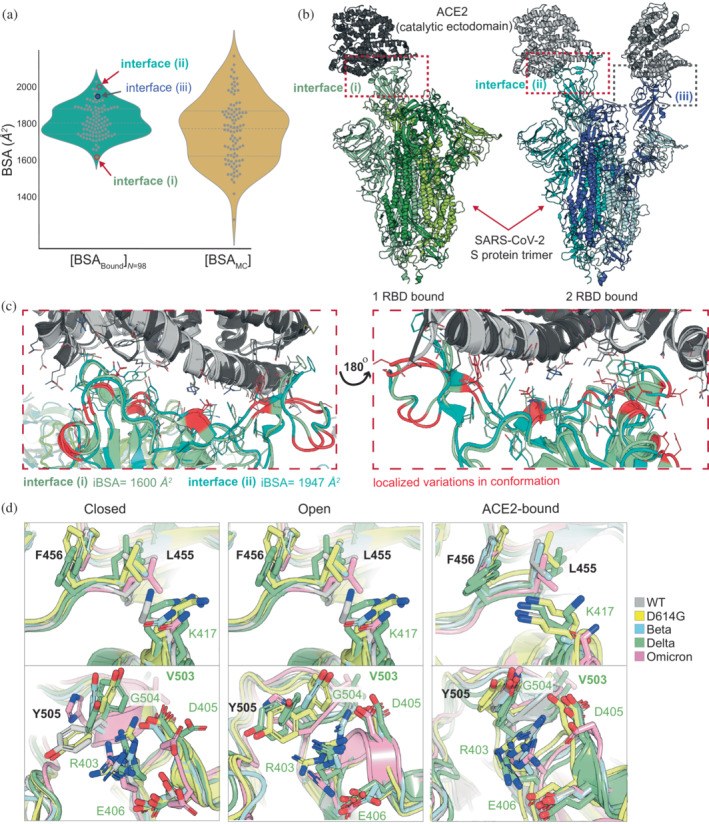
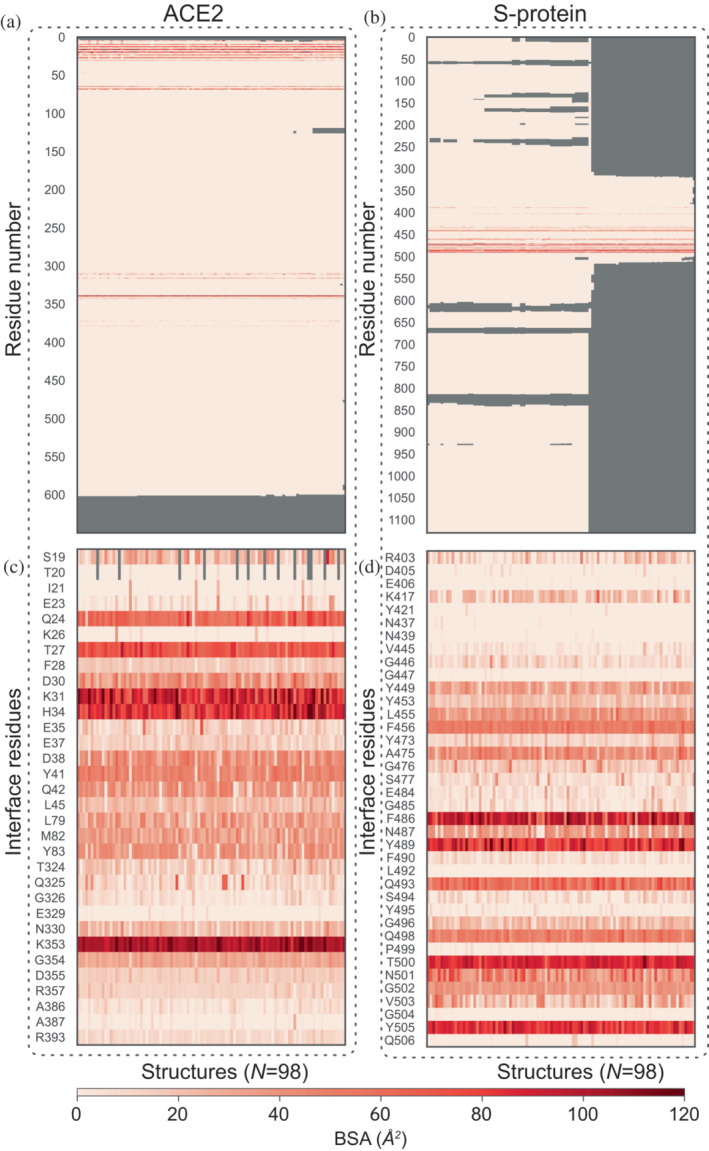
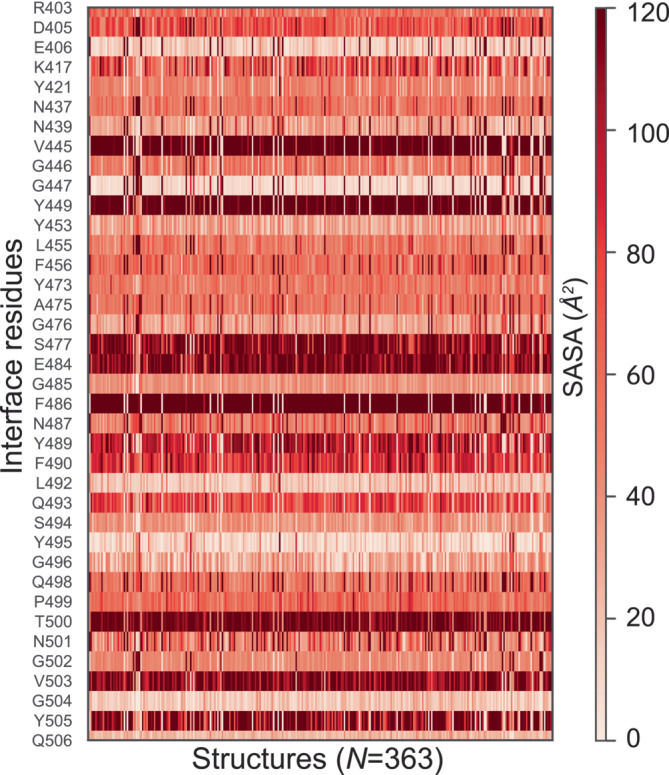
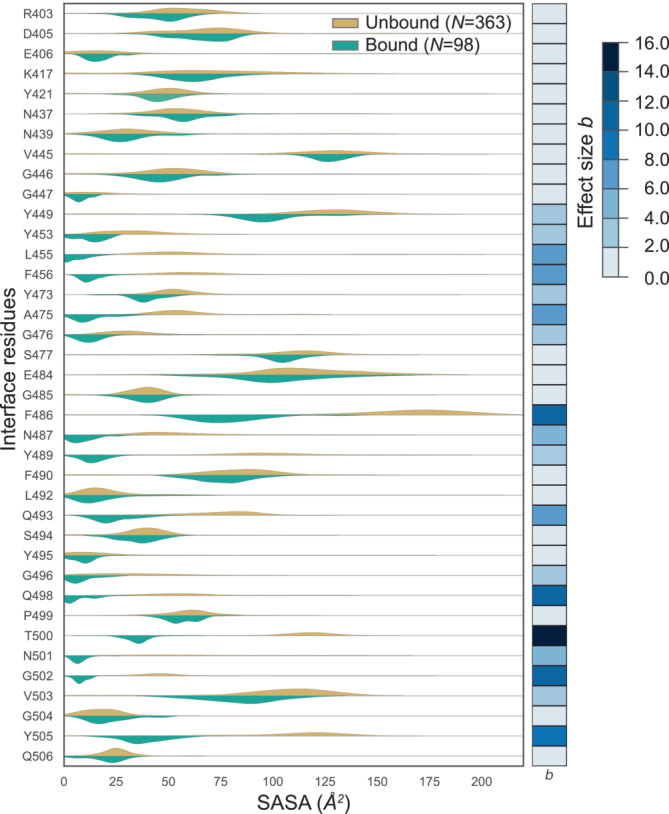
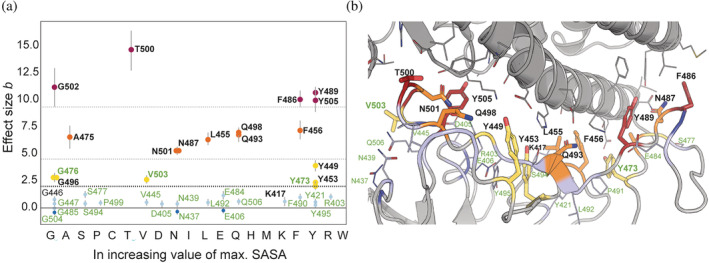
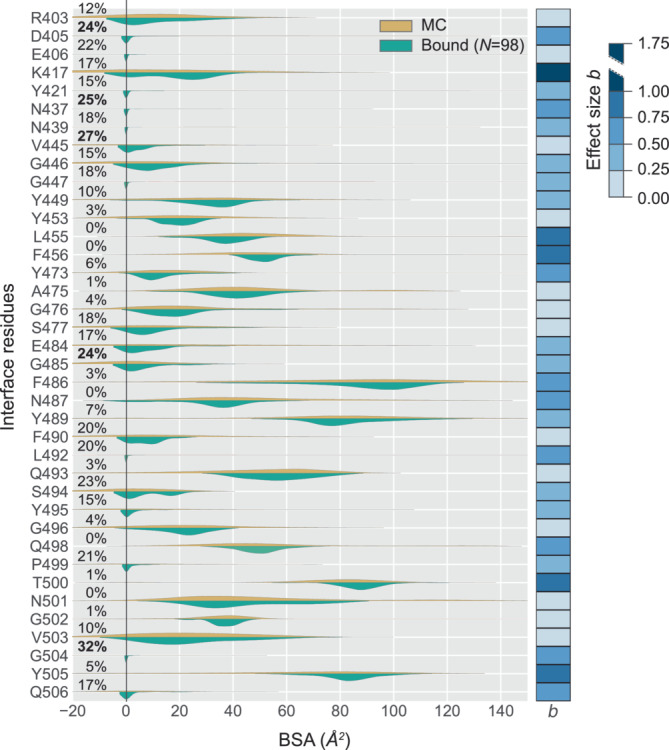
Understanding protein-protein interactions (PPIs) is fundamental to infer how different molecular systems work. A major component to model molecular recognition is the buried surface area (BSA), that is, the area that becomes inaccessible to solvent upon complex formation. To date, many attempts tried to connect BSA to molecular recognition principles, and in particular, to the underlying binding affinity. However, the most popular approach to calculate BSA is to use a single (or in some cases few) bound structures, consequently neglecting a wealth of structural information of the interacting proteins derived from ensembles corresponding to their unbound and bound states. Moreover, the most popular method inherently assumes the component proteins to bind as rigid entities. To address the above shortcomings, we developed a Monte Carlo method-based Interface Residue Assessment Algorithm (IRAA), to calculate a combined distribution of BSA for a given complex. Further, we apply our algorithm to human ACE2 and SARS-CoV-2 Spike protein complex, a system of prime importance. Results show a much broader distribution of BSA compared to that obtained from only the bound structure or structures and extended residue members of the interface with implications to the underlying biomolecular recognition. We derive that specific interface residues of ACE2 and of S-protein are consistently highly flexible, whereas other residues systematically show minor conformational variations. In effect, IRAA facilitates the use of all available structural data for any biomolecular complex of interest, extracting quantitative parameters with statistical significance, thereby providing a deeper biophysical understanding of the molecular system under investigation.
理解蛋白质-蛋白质相互作用(PPIs)对于推断不同分子系统的工作方式至关重要。建模分子识别的一个主要组成部分是埋置表面积(BSA),即在复合物形成后变得无法与溶剂接触的面积。迄今为止,许多尝试都试图将 BSA 与分子识别原理联系起来,特别是与潜在的结合亲和力联系起来。然而,计算 BSA 的最流行方法是使用单个(或在某些情况下少数)结合结构,因此忽略了大量来自对应于其未结合和结合状态的集合的相互作用蛋白质的结构信息。此外,最流行的方法本质上假设组成蛋白质以刚性实体结合。为了解决上述缺点,我们开发了一种基于蒙特卡罗方法的界面残基评估算法(IRAA),用于计算给定复合物的 BSA 的组合分布。此外,我们将我们的算法应用于人类 ACE2 和 SARS-CoV-2 Spike 蛋白复合物,这是一个非常重要的系统。结果表明,与仅从结合结构或结构获得的 BSA 分布相比,我们的算法得到的 BSA 分布要广泛得多,并扩展了界面的残基成员,这对潜在的生物分子识别有影响。我们得出结论,ACE2 和 S 蛋白的特定界面残基始终高度灵活,而其他残基则系统地表现出较小的构象变化。实际上,IRAA 促进了对任何感兴趣的生物分子复合物使用所有可用的结构数据,提取具有统计意义的定量参数,从而提供对所研究分子系统的更深层次的生物物理理解。