Laboratory of Human Cytogenetics, Molecular Genetics and Reproductive Biology, Farhat Hached University Hospital, Sousse, Tunisia.
Higher Institute of Biotechnology of Monastir, University of Monastir, Monastir, Tunisia.
PLoS One. 2022 Dec 1;17(12):e0278283. doi: 10.1371/journal.pone.0278283. eCollection 2022.
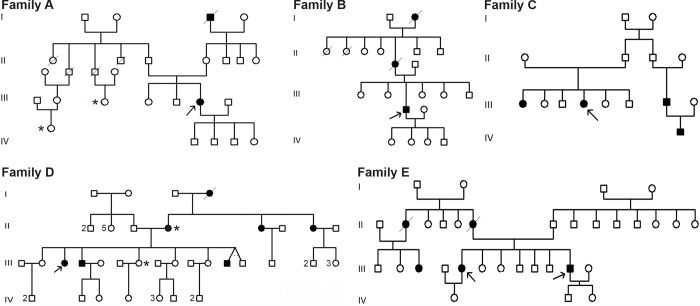
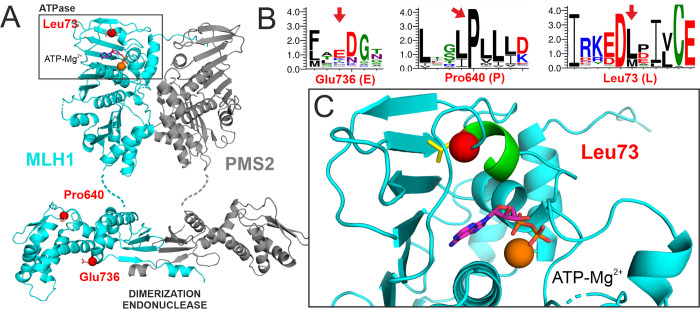
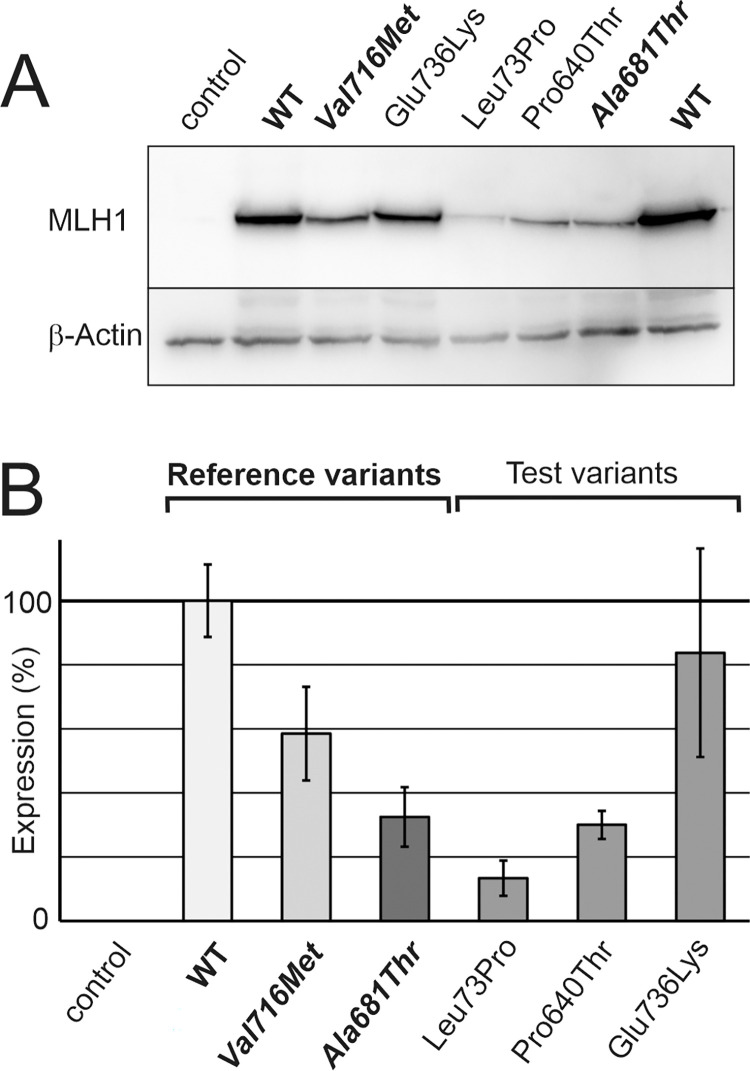
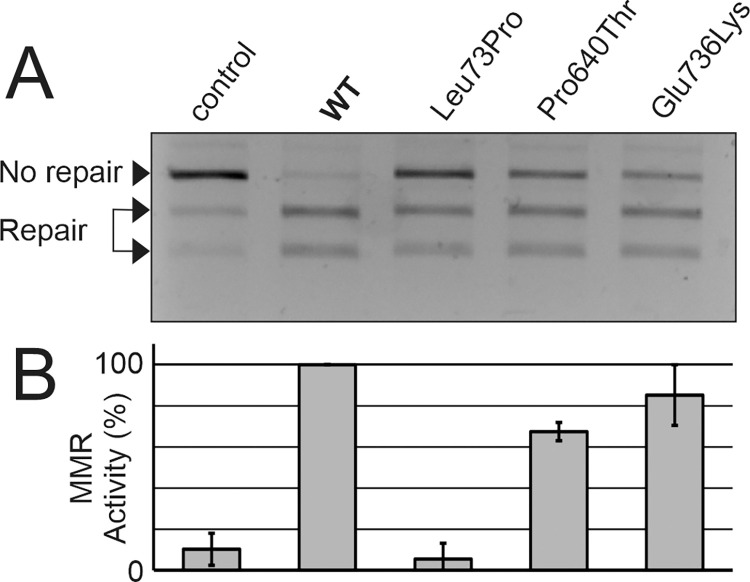
Lynch syndrome is a heritable condition caused by a heterozygous germline inactivating mutation of the DNA mismatch repair (MMR) genes, most commonly the MLH1 gene. However, one third of the identified alterations are missense variants, for which the clinical significance is unclear in many cases. We have identified three MLH1 missense alterations (p.(Glu736Lys), p.(Pro640Thr) and p.(Leu73Pro)) in six individuals from large Tunisian families. For none of these alterations, a classification of pathogenicity was available, consequently diagnosis, predictive testing and targeted surveillance in affected families was impossible. We therefore performed functional laboratory testing using a system testing stability as well as catalytic activity that includes clinically validated reference variants. Both p.(Leu73Pro) and p.(Pro640Thr) were found to be non-functional due to severe defects in protein stability and catalytic activity. In contrast, p.(Glu736Lys) was comparable to the wildtype protein and therefore considered a neutral substitution. Analysis of residue conservation and of the structural roles of the substituted residues corroborated these findings. In conjunction with the available clinical data, two variants fulfil classification criteria for class 4 "likely pathogenic". The findings of this work clarify the mechanism of pathogenicity of two unclear MLH1 variants and enables predictive testing and targeted surveillance in members of carrier families worldwide.
林奇综合征是一种遗传性疾病,由 DNA 错配修复 (MMR) 基因的杂合性胚系失活突变引起,最常见的是 MLH1 基因。然而,三分之一已确定的改变是错义变异,在许多情况下其临床意义尚不清楚。我们在六个来自大型突尼斯家族的个体中发现了三个 MLH1 错义改变(p.(Glu736Lys)、p.(Pro640Thr) 和 p.(Leu73Pro))。对于这些改变中的任何一个,都没有可用于致病性分类的信息,因此无法对受影响的家族进行诊断、预测性测试和靶向监测。因此,我们使用一种包括临床验证的参考变异体的系统测试稳定性和催化活性的功能实验室测试方法进行了研究。p.(Leu73Pro) 和 p.(Pro640Thr) 由于蛋白质稳定性和催化活性的严重缺陷而被发现是非功能性的。相比之下,p.(Glu736Lys) 与野生型蛋白相当,因此被认为是中性替换。对残基保守性和取代残基的结构作用的分析证实了这些发现。结合可用的临床数据,两种变体符合“可能致病性”类别 4 的分类标准。这项工作的结果阐明了两个不明 MLH1 变体的致病性机制,并使全世界携带者家族的预测性测试和靶向监测成为可能。