Translational Biobehavioral and Health Disparities Branch, Clinical Center, National Institutes of Health, Bethesda, MD, United States of America.
National Institute of Nursing Research, National Institutes of Health, Bethesda, MD, United States of America.
PLoS One. 2023 Jan 13;18(1):e0280293. doi: 10.1371/journal.pone.0280293. eCollection 2023.
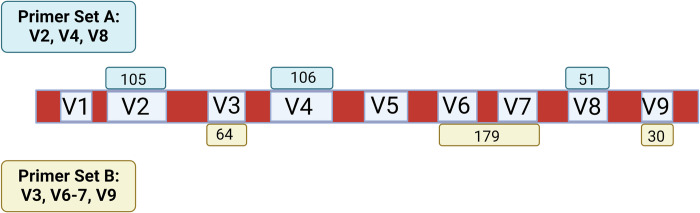
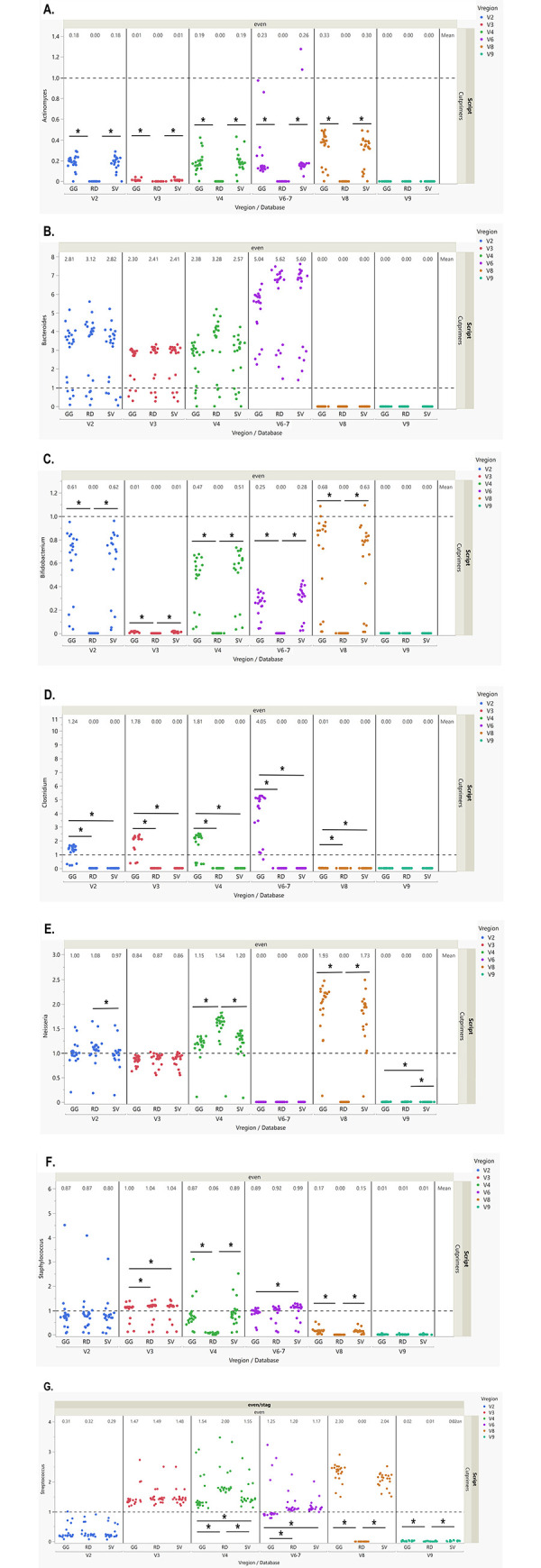
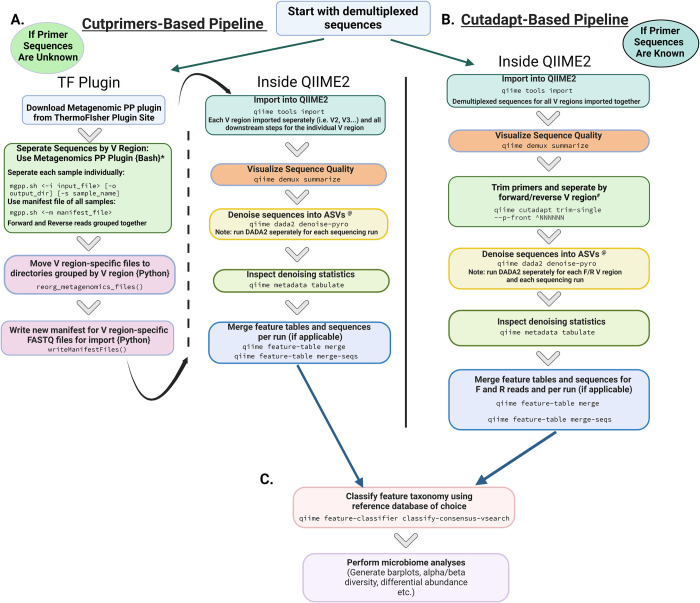
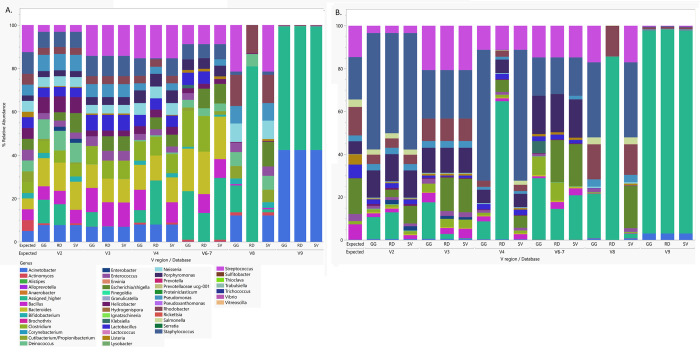
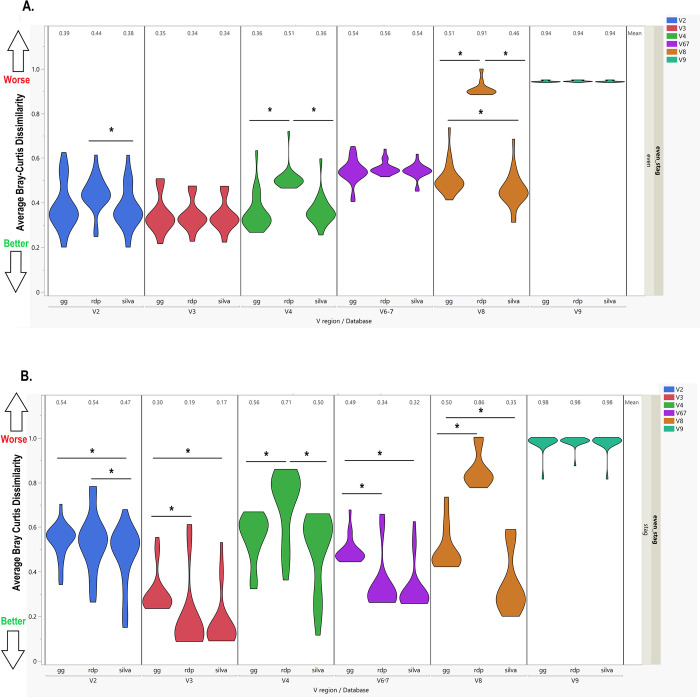
Microbiome research relies on next-generation sequencing and on downstream data analysis workflows. Several manufacturers have introduced multi-amplicon kits for microbiome characterization, improving speciation, but present unique challenges for analysis. The goal of this methodology study was to develop two analysis pipelines specific to mixed-orientation reads from multi-hypervariable (V) region amplicons. A secondary aim was to assess agreement with expected abundance, considering database and variable region. Mock community sequence data (n = 41) generated using the Ion16S™ Metagenomics Kit and Ion Torrent Sequencing Platform were analyzed using two workflows. Amplicons from V2, V3, V4, V6-7, V8 and V9 were deconvoluted using a specialized plugin based on CutPrimers. A separate workflow using Cutadapt is also presented. Three reference databases (Ribosomal Database Project, Greengenes and Silva) were used for taxonomic assignment. Bray-Curtis, Euclidean and Jensen-Shannon distance measures were used to evaluate overall annotation consistency, and specific taxon agreement was determined by calculating the ratio of observed to expected relative abundance. Reads that mapped to regions V2-V9 varied for both CutPrimers and Cutadapt-based methods. Within the CutPrimers-based pipeline, V3 amplicons had the best agreement with the expected distribution, tested using global distance measures, while V9 amplicons had the worst agreement. Accurate taxonomic annotation varied by genus-level taxon and V region analyzed. For the first time, we present a microbiome analysis pipeline that employs a specialized plugin to allow microbiome researchers to separate multi-amplicon data from the Ion16S Metagenomics Kit into V-specific reads. We also present an additional analysis workflow, modified for Ion Torrent mixed orientation reads. Overall, the global agreement of amplicons with the expected mock community abundances differed across V regions and reference databases. Benchmarking data should be referenced when planning a microbiome study to consider these biases related to sequencing and data analysis for multi-amplicon sequencing kits.
微生物组研究依赖于下一代测序和下游数据分析工作流程。几家制造商已经推出了用于微生物组特征描述的多扩增子试剂盒,提高了分类,但对分析提出了独特的挑战。本方法研究的目的是开发两种特定于多高变区 (V) 区扩增子的混合定向读取的分析管道。次要目标是考虑数据库和变量区,评估与预期丰度的一致性。使用 Ion16S™宏基因组试剂盒和 Ion Torrent 测序平台生成的模拟社区序列数据(n = 41)使用两种工作流程进行分析。使用基于 CutPrimers 的专用插件对 V2、V3、V4、V6-7、V8 和 V9 扩增子进行去卷积。还提出了一种使用 Cutadapt 的单独工作流程。使用三个参考数据库(核糖体数据库项目、Greengenes 和 Silva)进行分类分配。使用 Bray-Curtis、Euclidean 和 Jensen-Shannon 距离度量来评估整体注释一致性,通过计算观察到的相对丰度与预期相对丰度的比值来确定特定分类群的一致性。两种基于 CutPrimers 的方法的 V2-V9 映射读取都有所不同。在基于 CutPrimers 的管道中,使用全局距离度量测试时,V3 扩增子与预期分布的一致性最好,而 V9 扩增子的一致性最差。准确的分类注释因属级分类群和分析的 V 区而异。我们首次提出了一种微生物组分析管道,该管道使用专用插件允许微生物组研究人员将 Ion16S 宏基因组试剂盒的多扩增子数据分离成特定于 V 的读取。我们还提出了一种针对 Ion Torrent 混合定向读取的额外分析工作流程。总体而言,V 区和参考数据库的扩增子与预期模拟社区丰度的全球一致性存在差异。在计划微生物组研究时,应参考基准数据,以考虑与多扩增子测序试剂盒相关的测序和数据分析的这些偏差。