Gupta Deepali, Kumar Mukesh, Sharma Priyanka, Mohan Trishala, Prakash Amresh, Kumari Renu, Kaur Punit
Department of Biophysics, All India Institute of Medical Sciences, New Delhi 110026, India.
Division of Bio-Medical Informatics, Indian Council of Medical Research, New Delhi 110029, India.
Vaccines (Basel). 2022 Dec 22;11(1):23. doi: 10.3390/vaccines11010023.
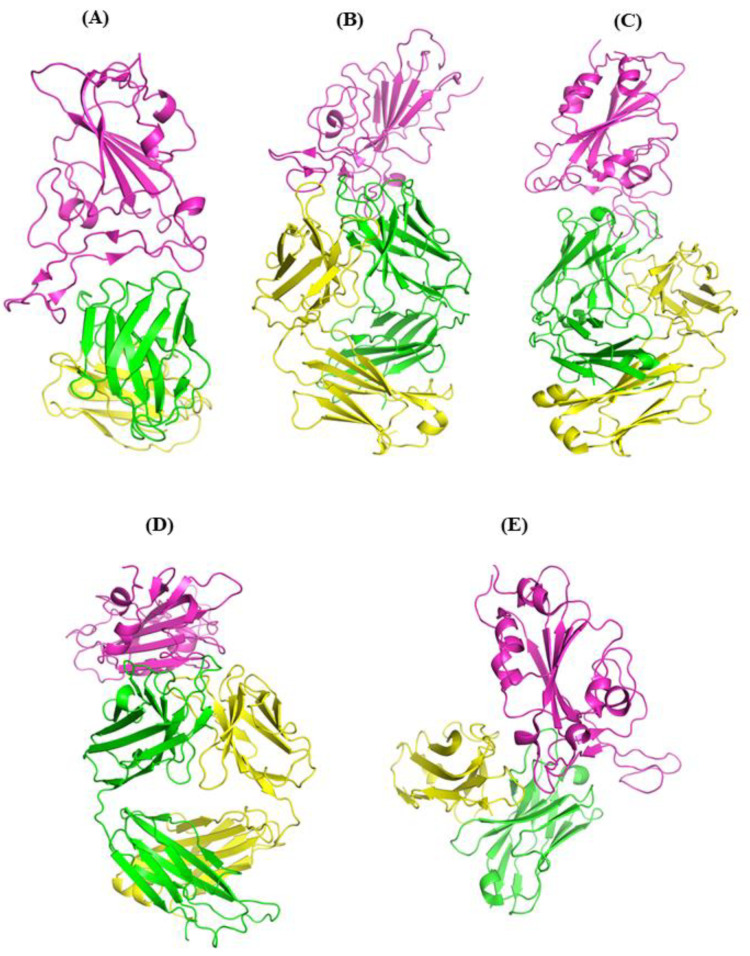
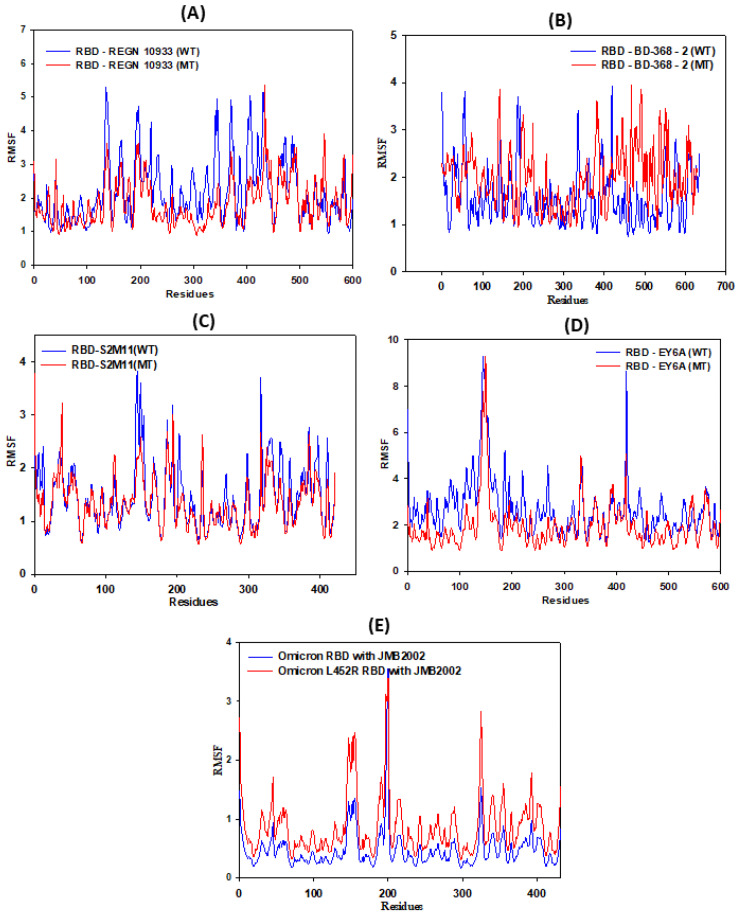
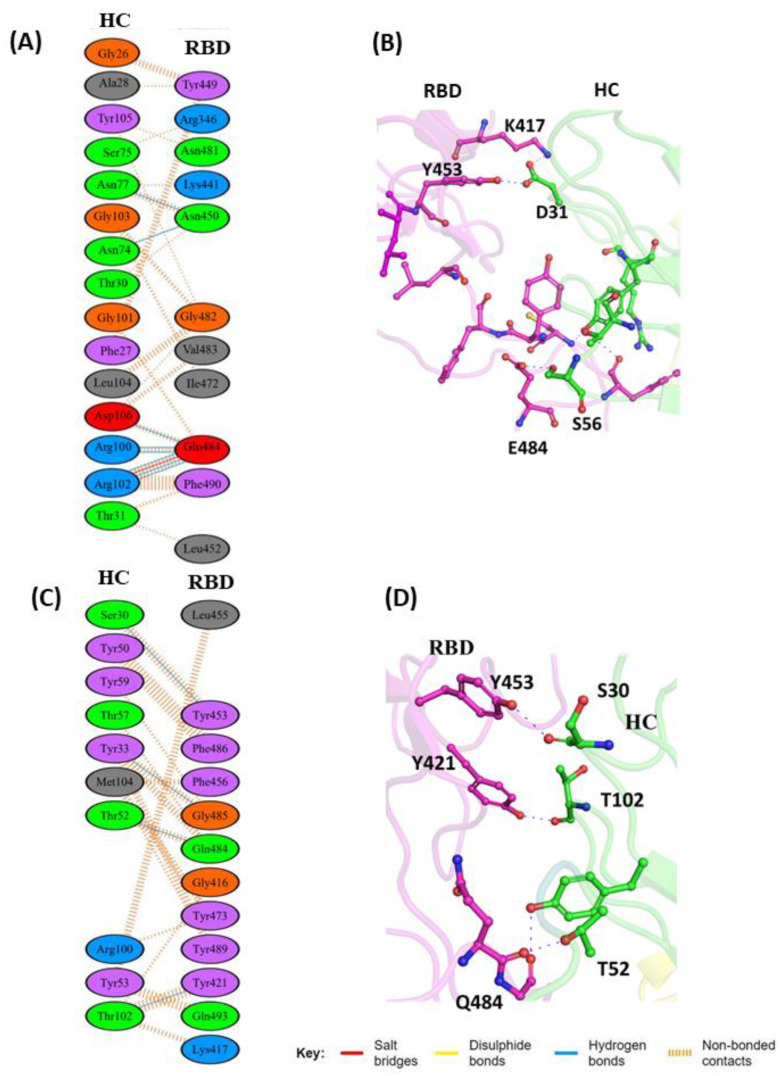
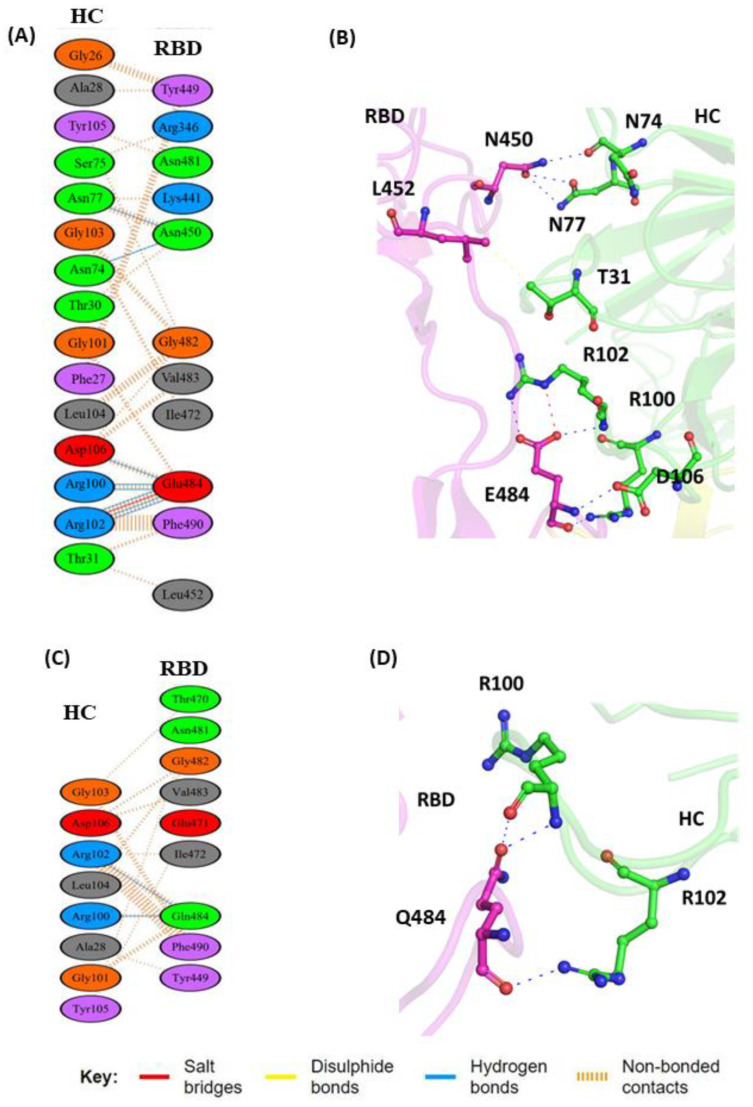
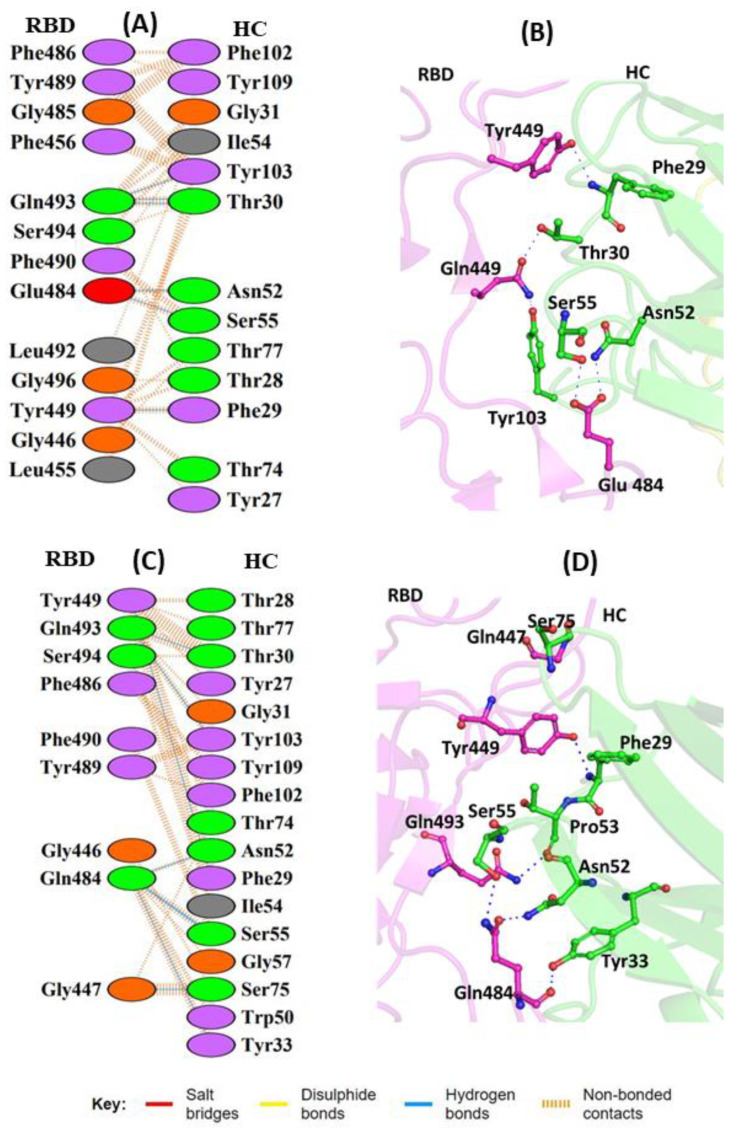
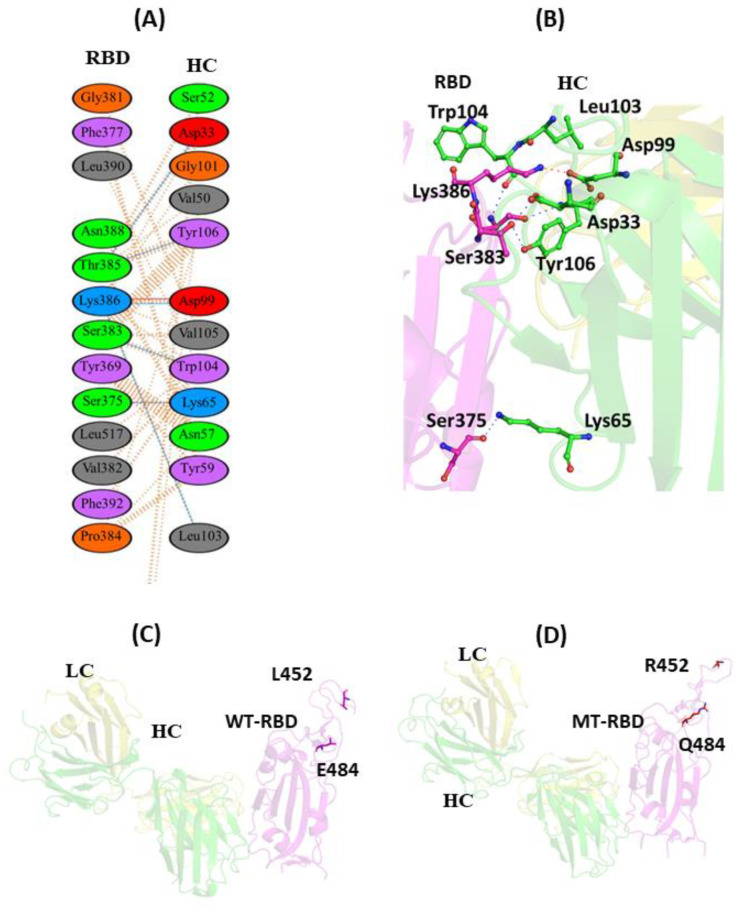
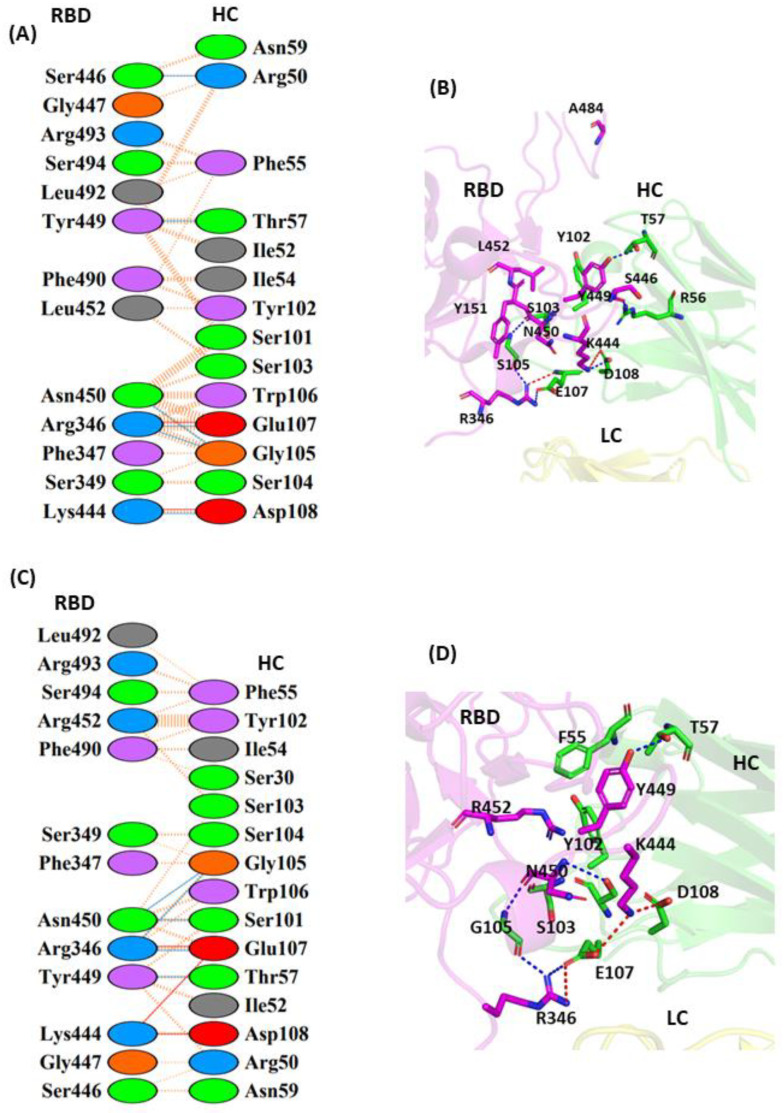
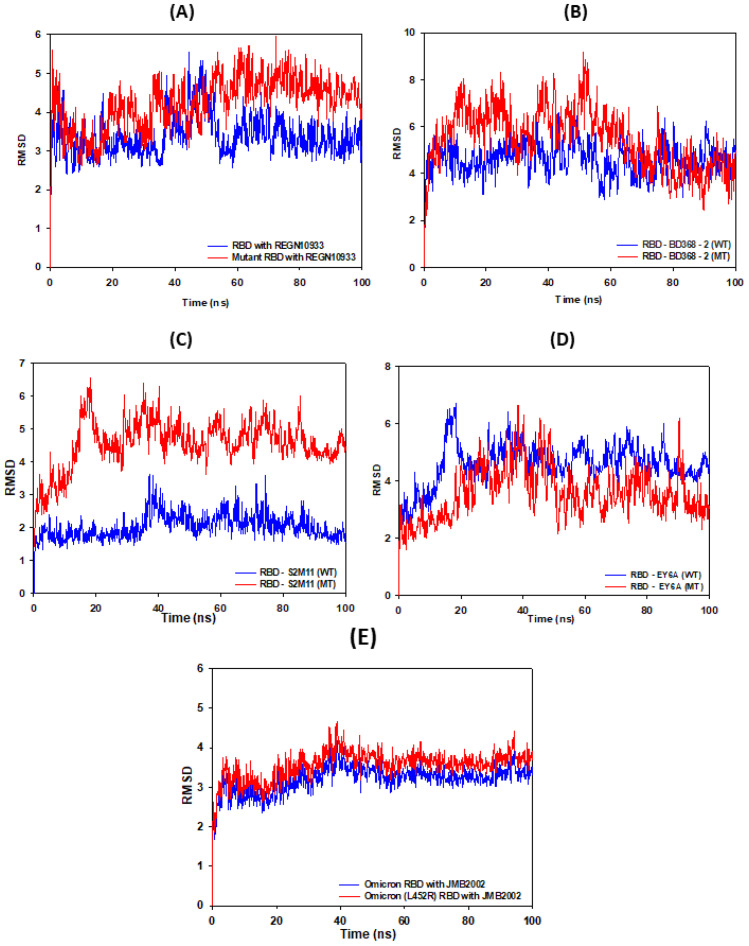
The COVID-19 pandemic, caused by SARS-CoV-2, emerges as a global health problem, as the viral genome is evolving rapidly to form several variants. Advancement and progress in the development of effective vaccines and neutralizing monoclonal antibodies are promising to combat viral infections. In the current scenario, several lineages containing "co-mutations" in the receptor-binding domain (RBD) region of the spike (S) protein are imposing new challenges. Co-occurrence of some co-mutations includes delta (L452R/T478K), kappa (L452R/E484Q), and a common mutation in both beta and gamma variants (E484K/N501Y). The effect of co-mutants (L452R/E484Q) on human angiotensin-converting enzyme 2 (hACE2) binding has already been elucidated. Here, for the first time, we investigated the role of these RBD co-mutations (L452R/E484Q) on the binding affinity of mAbs by adopting molecular dynamics (MD) simulation and free-energy binding estimation. The results obtained from our study suggest that the structural and dynamic changes introduced by these co-mutations reduce the binding affinity of the viral S protein to monoclonal antibodies (mAbs). The structural changes imposed by L452R create a charged patch near the interfacial surface that alters the affinity towards mAbs. In E484Q mutation, polar negatively charged E484 helps in the formation of electrostatic interaction, while the neutrally charged Q residue affects the interaction by forming repulsive forces. MD simulations along with molecular mechanics-generalized Born surface area (MMGBSA) studies revealed that the REGN 10933, BD-368-2, and S2M11 complexes have reduced binding affinity towards the double-mutant RBD. This indicates that their mutant (MT) structures have a stronger ability to escape from most antibodies than the wild type (WT). However, EY6A Ab showed higher affinity towards the double MT-RBD complex as compared to the WT. However, no significant effect of the per-residue contribution of double-mutated residues was observed, as this mAb does not interact with the region harboring L452 and E484 residues.
由严重急性呼吸综合征冠状病毒2(SARS-CoV-2)引起的2019冠状病毒病大流行已成为一个全球健康问题,因为病毒基因组正在迅速进化,形成了多个变种。有效疫苗和中和性单克隆抗体研发方面的进展有望对抗病毒感染。在当前情况下,刺突(S)蛋白受体结合域(RBD)区域包含“共突变”的几个谱系带来了新的挑战。一些共突变的共同出现包括德尔塔(L452R/T478K)、卡帕(L452R/E484Q),以及贝塔和伽马变种中的一个常见突变(E484K/N501Y)。共突变体(L452R/E484Q)对人血管紧张素转换酶2(hACE2)结合的影响已经得到阐明。在此,我们首次通过分子动力学(MD)模拟和自由能结合估计,研究了这些RBD共突变(L452R/E484Q)对单克隆抗体结合亲和力的作用。我们的研究结果表明,这些共突变引入的结构和动力学变化降低了病毒S蛋白与单克隆抗体(mAbs)的结合亲和力。L452R引起的结构变化在界面表面附近形成了一个带电区域,改变了对单克隆抗体的亲和力。在E484Q突变中,极性带负电的E484有助于形成静电相互作用,而带中性电荷的Q残基通过形成排斥力影响相互作用。MD模拟以及分子力学广义玻恩表面积(MMGBSA)研究表明,REGN 10933、BD-368-2和S2M11复合物对双突变RBD的结合亲和力降低。这表明它们的突变体(MT)结构比野生型(WT)具有更强的逃避大多数抗体的能力。然而,与野生型相比,EY6A抗体对双突变MT-RBD复合物表现出更高的亲和力。然而,未观察到双突变残基的每个残基贡献有显著影响,因为这种单克隆抗体不与含有L452和E484残基的区域相互作用。