Mester Rachel, Hou Kangcheng, Ding Yi, Meeks Gillian, Burch Kathryn S, Bhattacharya Arjun, Henn Brenna M, Pasaniuc Bogdan
Department of Computational Medicine, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA, 90095 USA.
Bioinformatics Interdepartmental Program, University of California, Los Angeles, Los Angeles, CA, 90095 USA.
bioRxiv. 2023 Jan 24:2023.01.20.524946. doi: 10.1101/2023.01.20.524946.
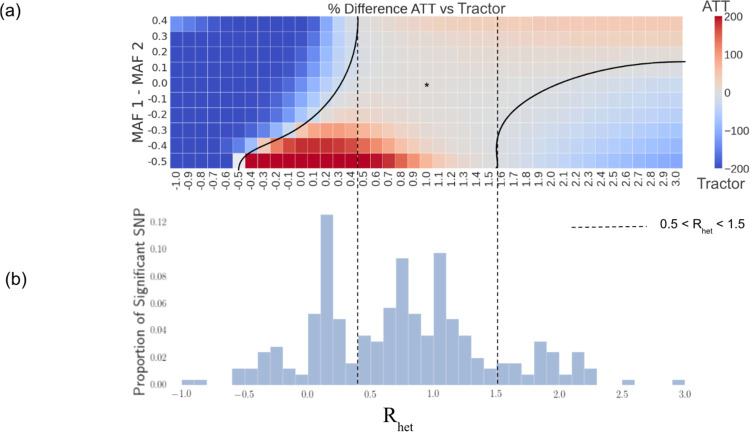
Genome-wide association studies (GWAS) have identified thousands of variants for disease risk. These studies have predominantly been conducted in individuals of European ancestries, which raises questions about their transferability to individuals of other ancestries. Of particular interest are admixed populations, usually defined as populations with recent ancestry from two or more continental sources. Admixed genomes contain segments of distinct ancestries that vary in composition across individuals in the population, allowing for the same allele to induce risk for disease on different ancestral backgrounds. This mosaicism raises unique challenges for GWAS in admixed populations, such as the need to correctly adjust for population stratification to balance type I error with statistical power. In this work we quantify the impact of differences in estimated allelic effect sizes for risk variants between ancestry backgrounds on association statistics. Specifically, while the possibility of estimated allelic effect-size heterogeneity by ancestry (HetLanc) can be modeled when performing GWAS in admixed populations, the extent of HetLanc needed to overcome the penalty from an additional degree of freedom in the association statistic has not been thoroughly quantified. Using extensive simulations of admixed genotypes and phenotypes we find that modeling HetLanc in its absence reduces statistical power by up to 72%. This finding is especially pronounced in the presence of allele frequency differentiation. We replicate simulation results using 4,327 African-European admixed genomes from the UK Biobank for 12 traits to find that for most significant SNPs HetLanc is not large enough for GWAS to benefit from modeling heterogeneity.
全基因组关联研究(GWAS)已经鉴定出数千个与疾病风险相关的变异。这些研究主要在欧洲血统的个体中进行,这引发了关于它们能否适用于其他血统个体的问题。特别令人感兴趣的是混合人群,通常定义为具有来自两个或更多大陆来源的近期祖先的人群。混合基因组包含不同祖先的片段,这些片段在人群中的个体组成上有所不同,使得相同的等位基因在不同的祖先背景下引发疾病风险。这种镶嵌性给混合人群中的GWAS带来了独特的挑战,例如需要正确调整群体分层以平衡I型错误和统计效力。在这项工作中,我们量化了祖先背景之间风险变异的估计等位基因效应大小差异对关联统计的影响。具体而言,虽然在混合人群中进行GWAS时可以对按祖先估计的等位基因效应大小异质性(HetLanc)的可能性进行建模,但克服关联统计中额外自由度带来的惩罚所需的HetLanc程度尚未得到充分量化。通过对混合基因型和表型进行广泛模拟,我们发现,在不存在HetLanc的情况下对其进行建模会使统计效力降低多达72%。这一发现在存在等位基因频率分化的情况下尤为明显。我们使用来自英国生物银行的4327个非洲 - 欧洲混合基因组对12个性状重复模拟结果,发现对于大多数显著的单核苷酸多态性(SNP),HetLanc不足以使GWAS从异质性建模中受益。