Gallardo Carmina, Casado Nadia, Soler Alejandro, Djadjovski Igor, Krivko Laura, Madueño Encarnación, Nieto Raquel, Perez Covadonga, Simon Alicia, Ivanova Emiliya, Donescu Daniel, Milicevik Vesna, Chondrokouki Eleni, Nurmoja Imbi, Frant Maciej, Feliziani Francesco, Václavek Petr, Pileviciene Simona, Marisa Arias
European Union Reference Laboratory for ASF (EURL-ASF): Centro De investigación en Sanidad Animal (CISA-INIA, CSIC), Madrid, Spain.
Faculty of Veterinary Medicine, University Ss. Cyril and Methodius in Skopje, Skopje, North Macedonia.
Front Vet Sci. 2023 Jan 25;10:1112850. doi: 10.3389/fvets.2023.1112850. eCollection 2023.
African swine fever (ASF) is a contagious viral disease of pigs and wild boar that poses a major threat to the global swine industry. The genotype II African swine fever virus (ASFV) entered the European Union (EU) in 2014 and since then fourteen countries have been affected, Italy and North Macedonia being the last in 2022. While whole genome sequencing remains the gold standard for the identification of new genetic markers, sequencing of multiple loci with significant variations could be used as a rapid and cost-effective alternative to track outbreaks and study disease evolution in endemic areas.
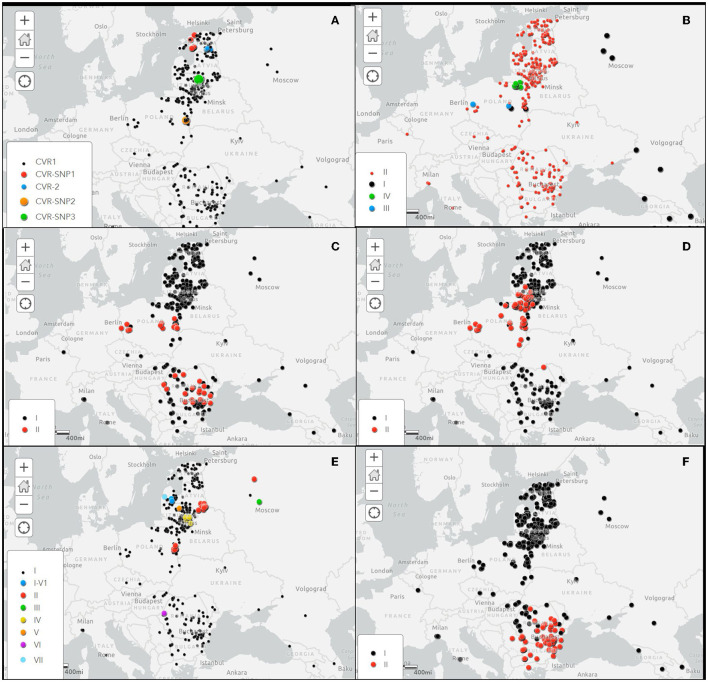
To further our understanding of the epidemiology and spread of ASFV in Europe, 382 isolates collected during 2007 to 2022 were sequenced. The study was initially performed by sequencing the central variable region (CVR), the intergenic region (IGR) between the and genes and the and genes. For further discrimination, two new PCRs were designed to amplify the IGR between the and genes of the multigene family 505 (MGF505) and the IGR between the and genes. The sequences obtained were compared with genotype II isolates from Europe and Asia.
The combination of the results obtained by sequencing these variable regions allowed to differentiate the European II-ASFV genotypes into 24 different groups. In addition, the SNP identified in the IGR - region, not previously described, grouped the viruses from North Macedonia that caused the 2022 outbreaks with viruses from Romania, Bulgaria, Serbia and Greece, differentiating from other genotype II isolates present in Europe and Asia. Furthermore, tandem repeat sequence (TRS) within the - genes of the multigene family 505 (MGF505) revealed eight different variants circulating.
These findings describe a new multi-gene approach sequencing method that can be used in routine genotyping to determine the origin of new introductions in ASF-free areas and track infection dynamics in endemic areas.
非洲猪瘟(ASF)是一种猪和野猪的传染性病毒病,对全球养猪业构成重大威胁。II型非洲猪瘟病毒(ASFV)于2014年进入欧盟(EU),自那时起已有14个国家受到影响,意大利和北马其顿是2022年受影响的最后两个国家。虽然全基因组测序仍然是鉴定新遗传标记的金标准,但对具有显著变异的多个位点进行测序可作为一种快速且经济高效的替代方法,用于追踪疫情爆发并研究流行地区的疾病演变。
为了进一步了解ASFV在欧洲的流行病学和传播情况,对2007年至2022年期间收集的382株分离株进行了测序。该研究最初通过对中央可变区(CVR)、 基因和 基因之间的基因间隔区(IGR)以及 基因和 基因进行测序来进行。为了进一步区分,设计了两个新的PCR来扩增多基因家族505(MGF505)的 基因和 基因之间的IGR以及 基因和 基因之间的IGR。将获得的序列与来自欧洲和亚洲的II型分离株进行比较。
通过对这些可变区进行测序获得的结果组合,能够将欧洲II型ASFV基因型分为24个不同的组。此外,在IGR - 区域中鉴定出的单核苷酸多态性(SNP)(此前未描述),将导致2022年疫情爆发的北马其顿病毒与来自罗马尼亚、保加利亚、塞尔维亚和希腊的病毒归为一组,与欧洲和亚洲存在的其他II型分离株区分开来。此外,多基因家族505(MGF505)的 基因内的串联重复序列(TRS)显示有八种不同的变体在传播。
这些发现描述了一种新的多基因方法测序方法,可用于常规基因分型,以确定无ASF地区新引入病毒的来源,并追踪流行地区的感染动态。