Departamento de Química Analítica, Química Física e Ingeniería Química, Universidad de Alcalá, Ctra. Madrid-Barcelona, Km 33,600, 28871 Alcalá de Henares, Madrid, Spain.
UMR 7019 LPCT, Université de Lorraine and CNRS, F-5400 Nancy, France.
Int J Mol Sci. 2023 Jan 28;24(3):2517. doi: 10.3390/ijms24032517.
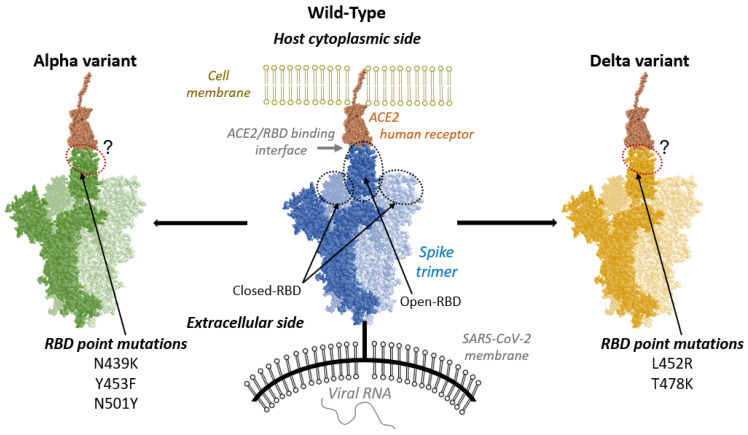
After a sudden and first spread of the pandemic caused by the novel SARS-CoV-2 (Severe Acute Respiratory Syndrome-Coronavirus 2) wild-type strain, mutants have emerged which have been associated with increased infectivity, inducing surges in the contagions. The first of the so-called variants of concerns, was firstly isolated in the United Kingdom and later renamed Alpha variant. Afterwards, in the middle of 2021, a new variant appeared called Delta. The latter is characterized by the presence of point mutations in the Spike protein of SARS-CoV-2, especially in the Receptor Binding Domain (RBD). When in its active conformation, the RBD can interact with the human receptor Angiotensin-Converting Enzyme 2 (ACE2) to allow the entry of the virions into cells. In this contribution, by using extended all-atom molecular dynamic simulations, complemented with machine learning post-processing, we analyze the changes in the molecular interaction network induced by these different strains in comparison with the wild-type. On one hand, although relevant variations are evidenced, only limited changes in the global stability indicators and in the flexibility profiles have been observed. On the other hand, key differences were obtained by tracking hydrophilic and hydrophobic molecular interactions, concerning both positioning at the ACE2/RBD interface and formation/disruption dynamic behavior.
在由新型 SARS-CoV-2(严重急性呼吸系统综合症-冠状病毒 2)野生型菌株引起的大流行突然首次传播后,出现了与传染性增加相关的突变体,导致感染激增。第一个所谓的关注变体,最初在英国分离出来,后来更名为 Alpha 变体。此后,在 2021 年年中,出现了一种新的变体,称为 Delta 变体。后者的特点是 SARS-CoV-2 的刺突蛋白(Spike protein)中存在点突变,特别是在受体结合域(Receptor Binding Domain,RBD)中。当处于其活性构象时,RBD 可以与人类受体血管紧张素转换酶 2(Angiotensin-Converting Enzyme 2,ACE2)相互作用,允许病毒粒子进入细胞。在本研究中,我们使用扩展的全原子分子动力学模拟,并辅以机器学习后处理,分析了这些不同菌株与野生型相比引起的分子相互作用网络的变化。一方面,尽管证据表明存在相关的变化,但仅观察到全局稳定性指标和柔韧性特征的有限变化。另一方面,通过跟踪亲水和疏水分子相互作用,包括在 ACE2/RBD 界面的定位以及形成/破坏动态行为,得到了关键差异。