School of Computer Science and Engineering, The Hebrew University of Jerusalem, Jerusalem, Israel.
Department of Genetics, The Institute of Life Sciences, The Hebrew University of Jerusalem, Jerusalem, Israel.
BMC Med Genomics. 2023 Feb 18;16(1):26. doi: 10.1186/s12920-023-01446-6.
The study of gene essentiality, which measures the importance of a gene for cell division and survival, is used for the identification of cancer drug targets and understanding of tissue-specific manifestation of genetic conditions. In this work, we analyze essentiality and gene expression data from over 900 cancer lines from the DepMap project to create predictive models of gene essentiality.
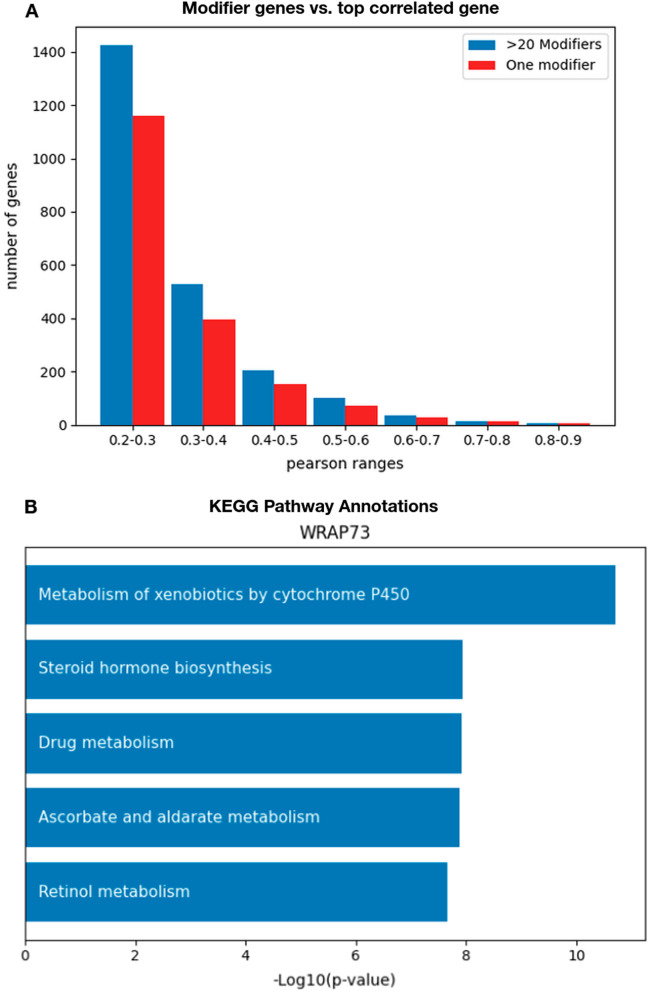
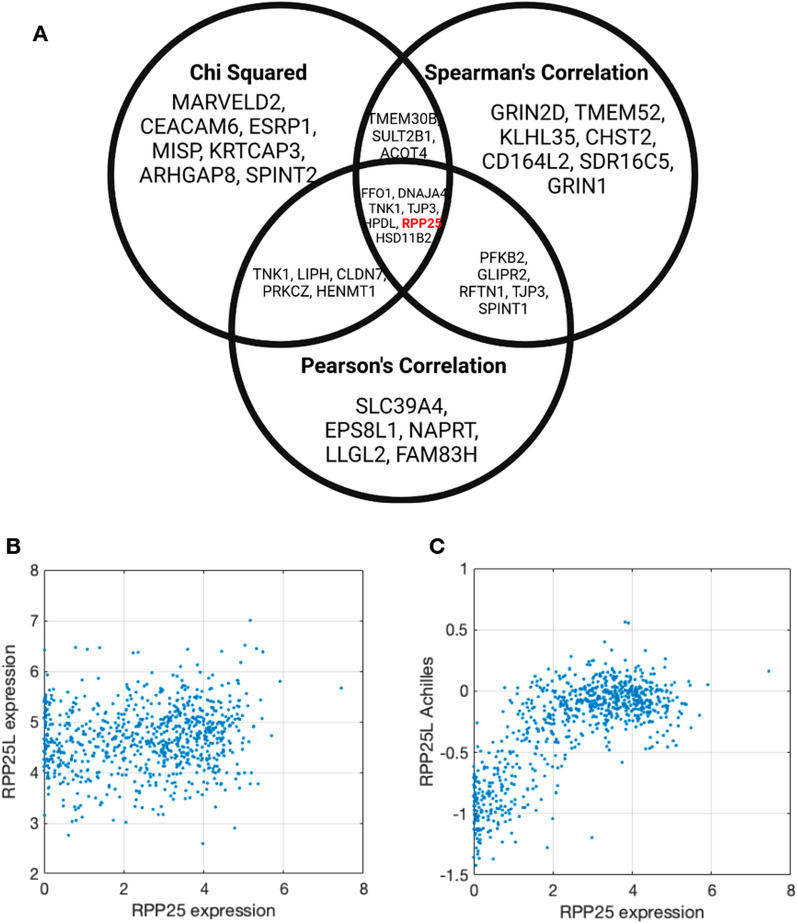
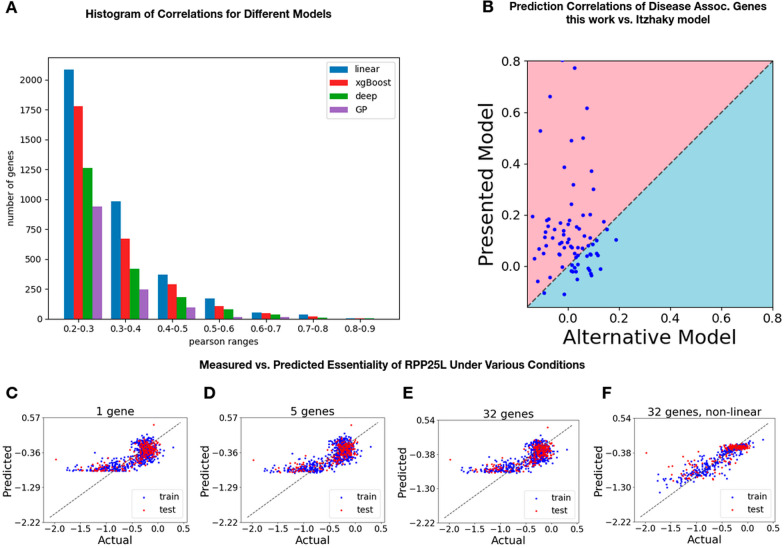
We developed machine learning algorithms to identify those genes whose essentiality levels are explained by the expression of a small set of "modifier genes". To identify these gene sets, we developed an ensemble of statistical tests capturing linear and non-linear dependencies. We trained several regression models predicting the essentiality of each target gene, and used an automated model selection procedure to identify the optimal model and hyperparameters. Overall, we examined linear models, gradient boosted trees, Gaussian process regression models, and deep learning networks.
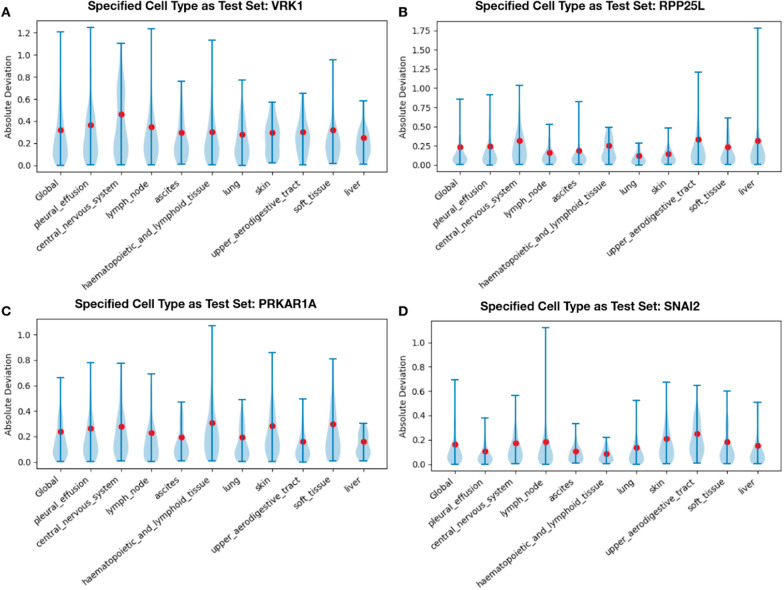
We identified nearly 3000 genes for which we accurately predict essentiality using gene expression data of a small set of modifier genes. We show that both in the number of genes we successfully make predictions for, as well as in the prediction accuracy, our model outperforms current state-of-the-art works.
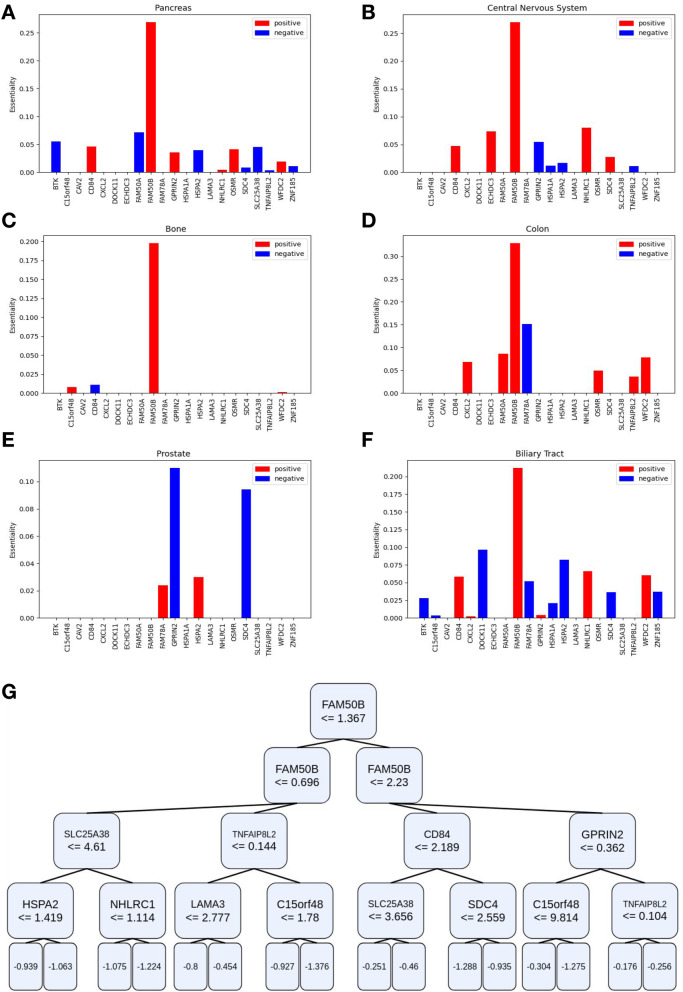
Our modeling framework avoids overfitting by identifying the small set of modifier genes, which are of clinical and genetic importance, and ignores the expression of noisy and irrelevant genes. Doing so improves the accuracy of essentiality prediction in various conditions and provides interpretable models. Overall, we present an accurate computational approach, as well as interpretable modeling of essentiality in a wide range of cellular conditions, thus contributing to a better understanding of the molecular mechanisms that govern tissue-specific effects of genetic disease and cancer.
基因必需性研究用于鉴定癌症药物靶点和理解遗传疾病在组织中的特异性表现,其衡量了基因对细胞分裂和存活的重要性。在这项工作中,我们分析了来自 DepMap 项目的 900 多个癌细胞系的必需性和基因表达数据,以创建基因必需性的预测模型。
我们开发了机器学习算法来识别那些其必需性水平由一小部分“修饰基因”的表达解释的基因。为了识别这些基因集,我们开发了一个捕捉线性和非线性依赖关系的统计测试集成。我们训练了几个回归模型来预测每个靶基因的必需性,并使用自动模型选择程序来识别最优模型和超参数。总体而言,我们检查了线性模型、梯度提升树、高斯过程回归模型和深度学习网络。
我们确定了近 3000 个基因,我们可以使用一小部分修饰基因的表达数据准确预测这些基因的必需性。我们表明,无论是成功预测的基因数量,还是预测准确性,我们的模型都优于当前的最新技术。
我们的建模框架通过识别具有临床和遗传重要性的少量修饰基因,避免了过度拟合,同时忽略了嘈杂和不相关基因的表达。这样做提高了各种条件下必需性预测的准确性,并提供了可解释的模型。总的来说,我们提出了一种准确的计算方法,以及在广泛的细胞条件下对必需性的可解释建模,从而有助于更好地理解控制遗传疾病和癌症组织特异性效应的分子机制。