Bioinformatics and Computational Biology Program, Iowa State University, Ames, IA 50011.
Roy J. Carver Department of Biochemistry, Biophysics and Molecular Biology, Iowa State University, Ames, IA 50011.
Proc Natl Acad Sci U S A. 2023 Feb 28;120(9):e2211823120. doi: 10.1073/pnas.2211823120. Epub 2023 Feb 24.
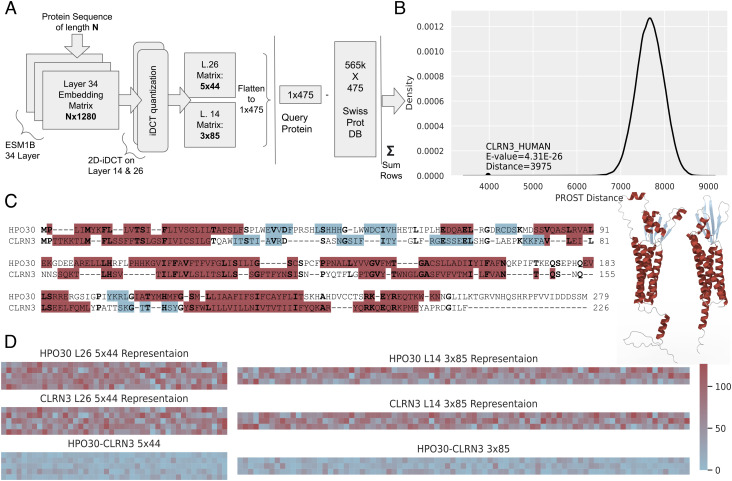
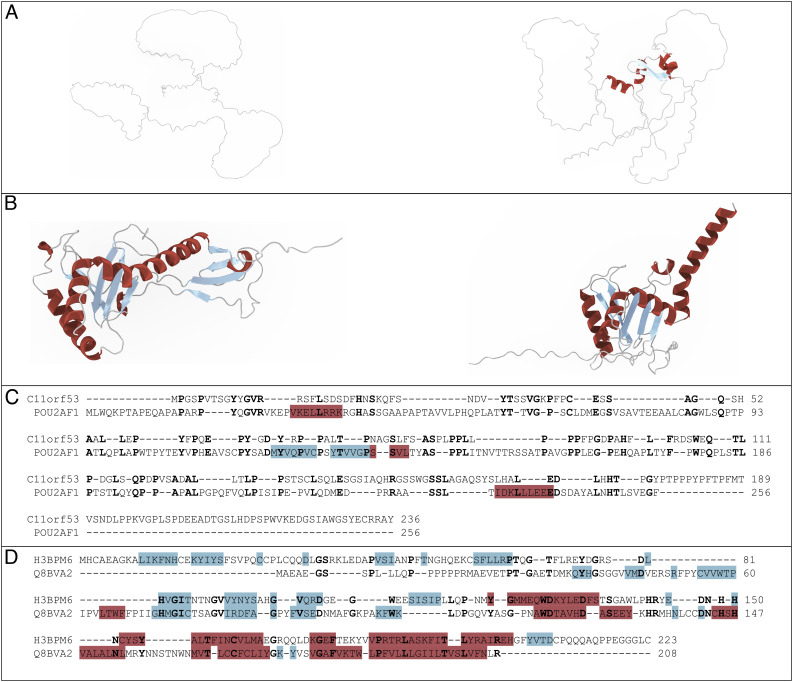
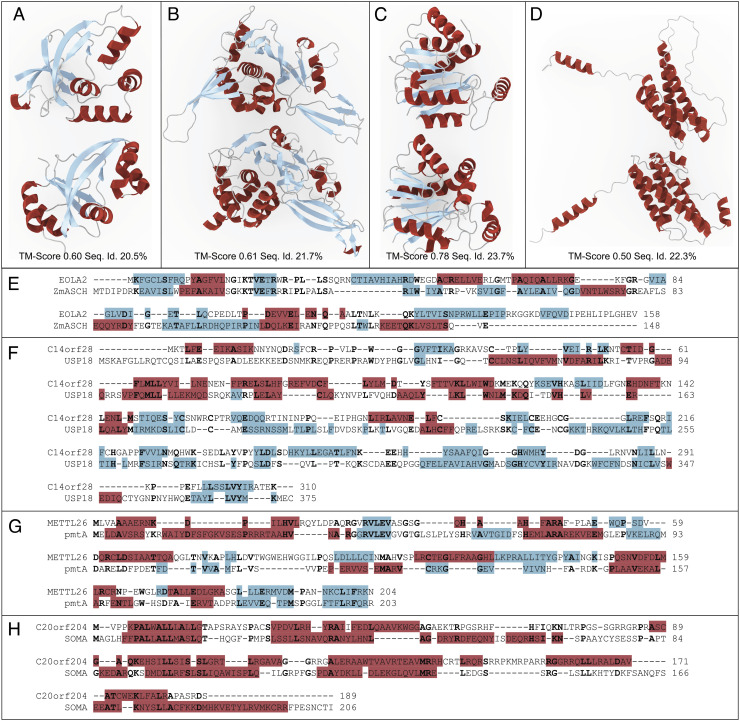
There are several hundred million protein sequences, but the relationships among them are not fully available from existing homolog detection methods. There is an essential need for an improved method to push homolog detection to lower levels of sequence identity. The method used here relies on a language model to represent proteins numerically in a matrix (an embedding) and uses discrete cosine transforms to compress the data to extract the most essential part, significantly reducing the data size. This PRotein Ortholog Search Tool (PROST) is significantly faster with linear runtimes, and most importantly, computes the distances between pairs of protein sequences to yield homologs at significantly lower levels of sequence identity than previously. The extent of allosteric effects in proteins points out the importance of global aspects of structure and sequence. PROST excels at global homology detection but not at detecting local homologs. Results are validated by strong similarities between the corresponding pairs of structures. The number of remote homologs detected increased significantly and pushes the effective sequence matches more deeply into the twilight zone. Human protein sequences presently having no assigned function now find significant numbers of putative homologs for 93% of cases and structurally verified assigned functions for 76.4% of these cases. The data compression enables massive searches for homologs with short search times while yielding significant gains in the numbers of remote homologs detected. The method is sufficiently efficient to permit whole-genome/proteome comparisons. The PROST web server is accessible at https://mesihk.github.io/prost.
有数亿种蛋白质序列,但现有的同源检测方法无法完全揭示它们之间的关系。因此,我们迫切需要一种改进的方法,将同源检测推进到更低的序列同一性水平。这里使用的方法依赖于语言模型将蛋白质数值表示为矩阵(嵌入),并使用离散余弦变换来压缩数据以提取最基本的部分,从而大大减少数据量。这种 PRotein Ortholog Search Tool (PROST) 不仅运行时间呈线性,而且最重要的是,它计算了蛋白质序列对之间的距离,从而以比以前更低的序列同一性水平产生了同源物。蛋白质的变构效应程度指出了结构和序列全局方面的重要性。PROST 在全局同源检测方面表现出色,但在检测局部同源物方面却不尽如人意。结果通过对应结构之间的强相似性得到验证。检测到的远程同源物数量显著增加,并将有效序列匹配推向更深的黄昏区域。目前没有指定功能的人类蛋白质序列现在在 93%的情况下发现了大量假定的同源物,在这些情况下的 76.4%具有结构上验证的指定功能。数据压缩使得可以在短时间内进行大规模的同源搜索,同时大大增加了检测到的远程同源物数量。该方法效率足够高,允许进行全基因组/蛋白质组比较。PROST 网络服务器可在 https://mesihk.github.io/prost 上访问。