Kobilka Institute of Innovative Drug Discovery, School of Medicine, The Chinese University of Hong Kong (Shenzhen), Shenzhen 518172, China.
Center of Data Mining and Biomedical informatics, Faculty of Medical Technology, Mahidol University, Bangkok 10700, Thailand.
Molecules. 2023 Feb 9;28(4):1679. doi: 10.3390/molecules28041679.
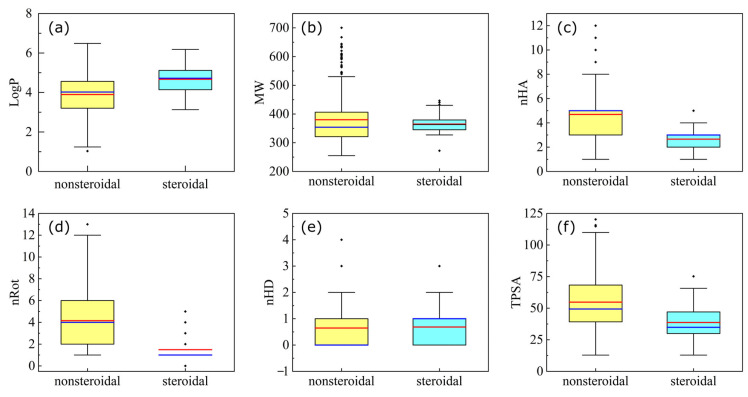
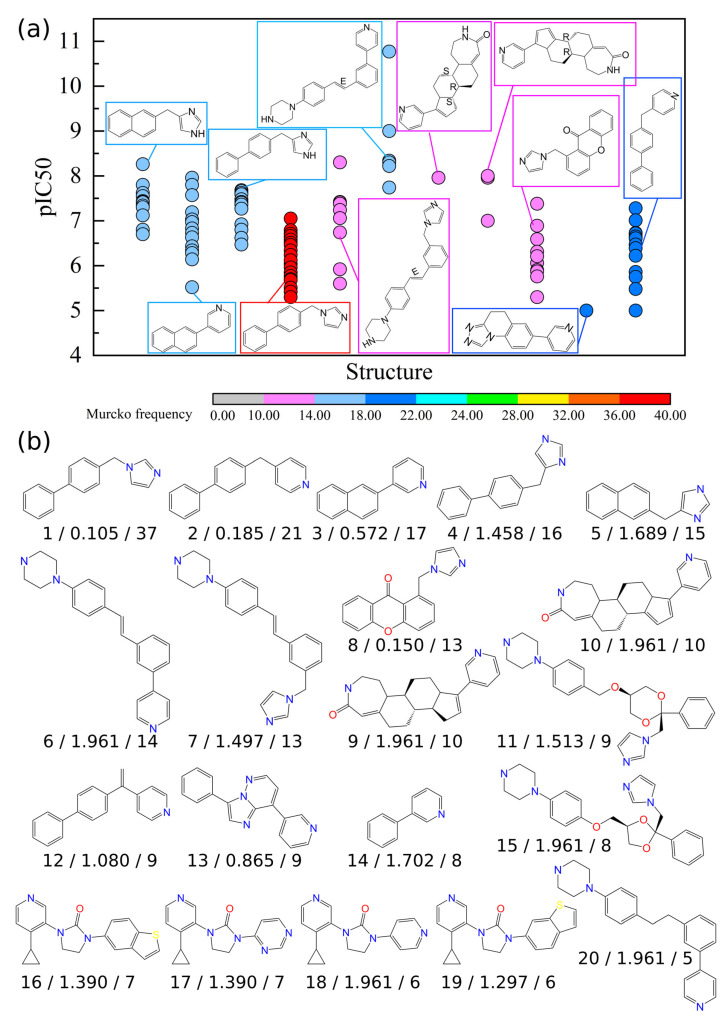
Cytochrome P450 17A1 (CYP17A1) is one of the key enzymes in steroidogenesis that produces dehydroepiandrosterone (DHEA) from cholesterol. Abnormal DHEA production may lead to the progression of severe diseases, such as prostatic and breast cancers. Thus, CYP17A1 is a druggable target for anti-cancer molecule development. In this study, cheminformatic analyses and quantitative structure-activity relationship (QSAR) modeling were applied on a set of 962 CYP17A1 inhibitors (i.e., consisting of 279 steroidal and 683 nonsteroidal inhibitors) compiled from the ChEMBL database. For steroidal inhibitors, a QSAR classification model built using the PubChem fingerprint along with the extra trees algorithm achieved the best performance, reflected by the accuracy values of 0.933, 0.818, and 0.833 for the training, cross-validation, and test sets, respectively. For nonsteroidal inhibitors, a systematic cheminformatic analysis was applied for exploring the chemical space, Murcko scaffolds, and structure-activity relationships (SARs) for visualizing distributions, patterns, and representative scaffolds for drug discoveries. Furthermore, seven total QSAR classification models were established based on the nonsteroidal scaffolds, and two activity cliff (AC) generators were identified. The best performing model out of these seven was model VIII, which is built upon the PubChem fingerprint along with the random forest algorithm. It achieved a robust accuracy across the training set, the cross-validation set, and the test set, i.e., 0.96, 0.92, and 0.913, respectively. It is anticipated that the results presented herein would be instrumental for further CYP17A1 inhibitor drug discovery efforts.
细胞色素 P450 17A1(CYP17A1)是类固醇生成中的关键酶之一,可将胆固醇转化为脱氢表雄酮(DHEA)。DHEA 产生异常可能导致前列腺癌和乳腺癌等严重疾病的进展。因此,CYP17A1 是开发抗癌分子的可用药靶标。在这项研究中,应用化学信息学分析和定量构效关系(QSAR)建模对一组 962 种 CYP17A1 抑制剂(即由 279 种甾体和 683 种非甾体抑制剂组成)进行了研究,这些抑制剂来自 ChEMBL 数据库。对于甾体抑制剂,使用 PubChem 指纹和 ExtraTrees 算法构建的 QSAR 分类模型表现最佳,其训练集、交叉验证集和测试集的准确率分别为 0.933、0.818 和 0.833。对于非甾体抑制剂,应用系统化学信息学分析探索化学空间、Murcko 支架和结构-活性关系,以可视化分布、模式和代表性支架,用于药物发现。此外,基于非甾体支架建立了七个总 QSAR 分类模型,并确定了两个活性悬崖(AC)生成器。这七个模型中表现最好的是模型 VIII,它是基于 PubChem 指纹和随机森林算法构建的。它在训练集、交叉验证集和测试集上均表现出稳健的准确性,分别为 0.96、0.92 和 0.913。预计本文的结果将有助于进一步的 CYP17A1 抑制剂药物发现工作。