Prajapati Milankumar, Zhang Jared Z, Mercadante Courtney J, Kowalski Heather L, Delaney Bradley, Anderson Jessica A, Guo Shuling, Aghajan Mariam, Bartnikas Thomas B
Department of Pathology and Laboratory Medicine, Brown University, Providence, Rhode Island, 02912, USA.
Ionis Pharmaceuticals, Inc., Carlsbad, CA, 92010, USA.
bioRxiv. 2023 Feb 21:2023.02.20.529270. doi: 10.1101/2023.02.20.529270.
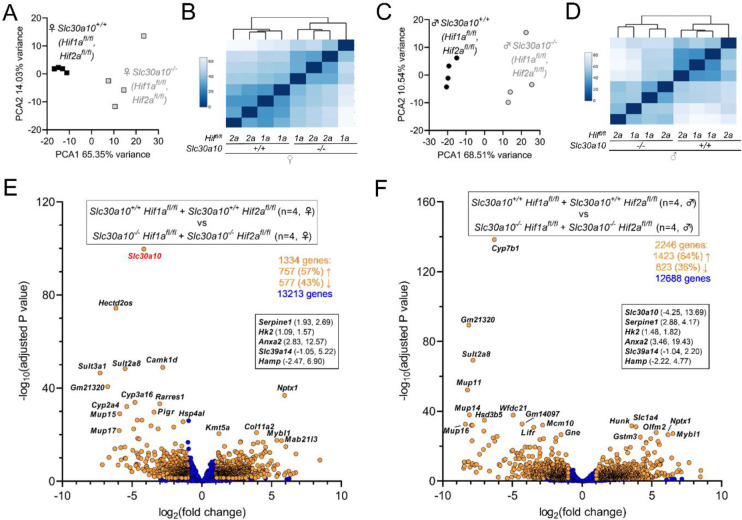
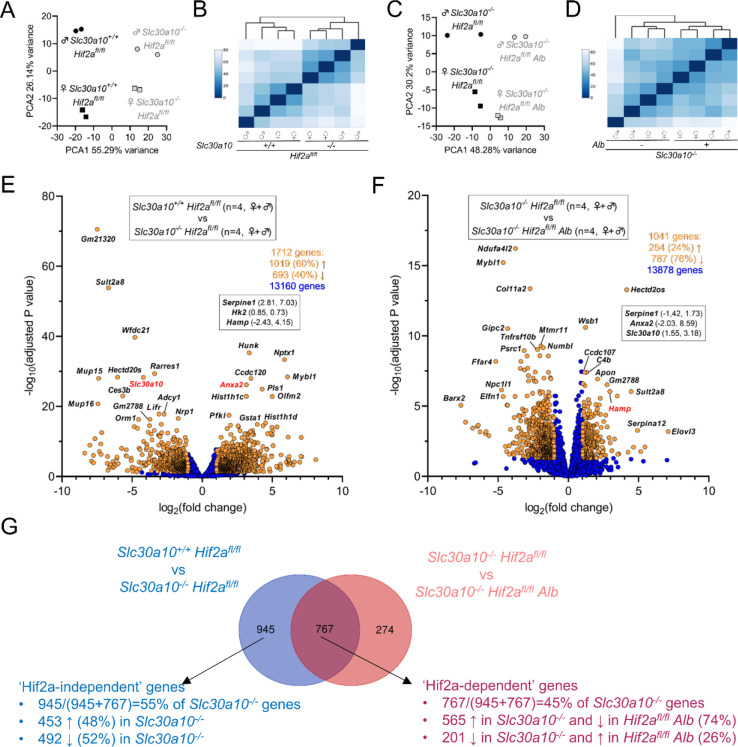
Manganese is an essential yet potentially toxic metal. Initially reported in 2012, mutations in SLC30A10 are the first known inherited cause of manganese excess. SLC30A10 is an apical membrane transport protein that exports manganese from hepatocytes into bile and from enterocytes into the lumen of the gastrointestinal tract. SLC30A10 deficiency results in impaired gastrointestinal manganese excretion, leading to severe manganese excess, neurologic deficits, liver cirrhosis, polycythemia, and erythropoietin excess. Neurologic and liver disease are attributed to manganese toxicity. Polycythemia is attributed to erythropoietin excess, but the basis of erythropoietin excess in SLC30A10 deficiency has yet to be established. Here we demonstrate that erythropoietin expression is increased in liver but decreased in kidneys in Slc30a10-deficient mice. Using pharmacologic and genetic approaches, we show that liver expression of hypoxia-inducible factor 2 (Hif2), a transcription factor that mediates the cellular response to hypoxia, is essential for erythropoietin excess and polycythemia in Slc30a10-deficient mice, while hypoxia-inducible factor 1 (HIF1) plays no discernible role. RNA-seq analysis determined that Slc30a10-deficient livers exhibit aberrant expression of a large number of genes, most of which align with cell cycle and metabolic processes, while hepatic Hif2 deficiency attenuates differential expression of half of these genes in mutant mice. One such gene downregulated in Slc30a10-deficient mice in a Hif2-dependent manner is hepcidin, a hormonal inhibitor of dietary iron absorption. Our analyses indicate that hepcidin downregulation serves to increase iron absorption to meet the demands of erythropoiesis driven by erythropoietin excess. Finally, we also observed that hepatic Hif2 deficiency attenuates tissue manganese excess, although the underlying cause of this observation is not clear at this time. Overall, our results indicate that HIF2 is a key determinant of pathophysiology in SLC30A10 deficiency.
锰是一种必需但具有潜在毒性的金属。2012年首次报道,SLC30A10基因突变是已知的首个遗传性锰过量病因。SLC30A10是一种顶端膜转运蛋白,可将锰从肝细胞转运至胆汁,并从肠细胞转运至胃肠道管腔。SLC30A10缺乏会导致胃肠道锰排泄受损,进而导致严重的锰过量、神经功能缺损、肝硬化、红细胞增多症和促红细胞生成素过量。神经和肝脏疾病归因于锰中毒。红细胞增多症归因于促红细胞生成素过量,但SLC30A10缺乏时促红细胞生成素过量的基础尚未明确。在此,我们证明Slc30a10基因缺陷小鼠肝脏中促红细胞生成素表达增加,而肾脏中促红细胞生成素表达减少。通过药理学和遗传学方法,我们表明缺氧诱导因子2(Hif2)的肝脏表达对于Slc30a10基因缺陷小鼠的促红细胞生成素过量和红细胞增多症至关重要,Hif2是一种介导细胞对缺氧反应的转录因子,而缺氧诱导因子1(HIF1)则没有明显作用。RNA测序分析确定,Slc30a10基因缺陷的肝脏表现出大量基因的异常表达,其中大多数与细胞周期和代谢过程相关,而肝脏Hif2缺乏会减弱突变小鼠中这些基因一半的差异表达。在Slc30a10基因缺陷小鼠中以Hif2依赖方式下调的一个这样的基因是铁调素,它是膳食铁吸收的激素抑制剂。我们的分析表明,铁调素下调有助于增加铁吸收,以满足促红细胞生成素过量驱动的红细胞生成需求。最后,我们还观察到肝脏Hif2缺乏会减轻组织锰过量,尽管目前尚不清楚这一观察结果的潜在原因。总体而言,我们的结果表明HIF2是SLC30A10缺乏病理生理学的关键决定因素。