Harley Peter, Paredes-Redondo Amaia, Grenci Gianluca, Viasnoff Virgile, Lin Yung-Yao, Lieberam Ivo
Centre for Gene Therapy & Regenerative Medicine, Kings College London, London SE1 9RT, UK.
Centre for Developmental Neurobiology and MRC Centre for Neurodevelopmental Disorders, Institute of Psychiatry, Psychology and Neuroscience, Kings College London, London SE1 1UL, UK.
Bio Protoc. 2023 Mar 5;13(5):e4624. doi: 10.21769/BioProtoc.4624.
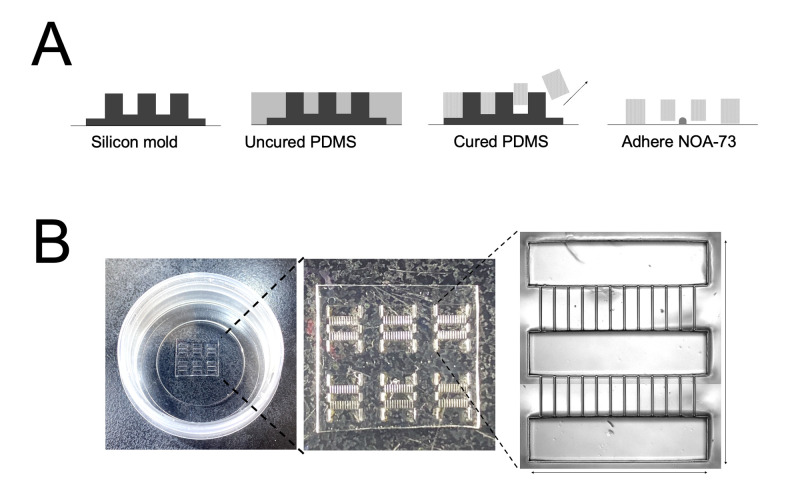
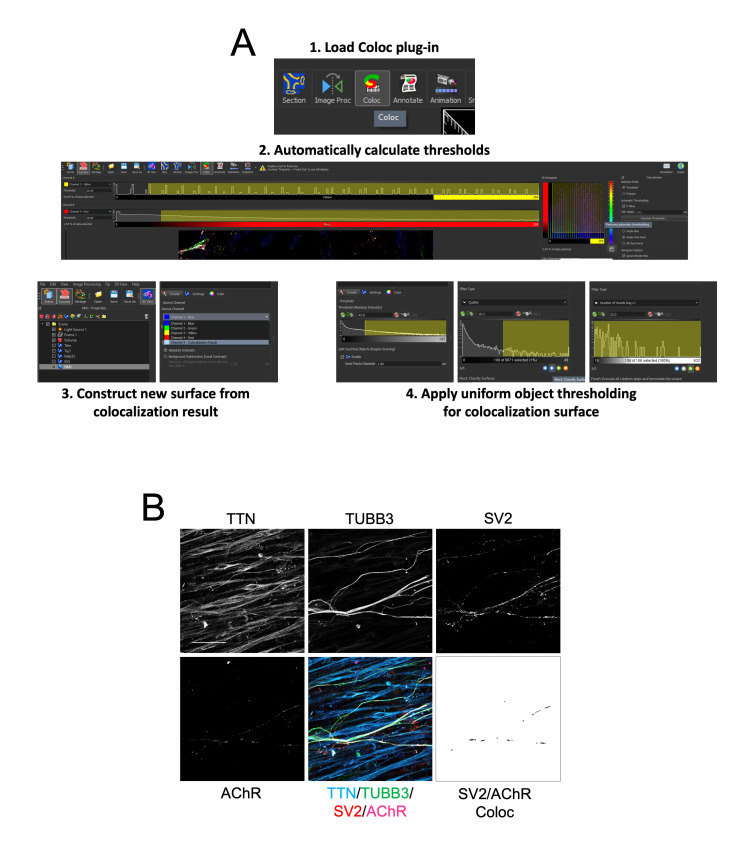
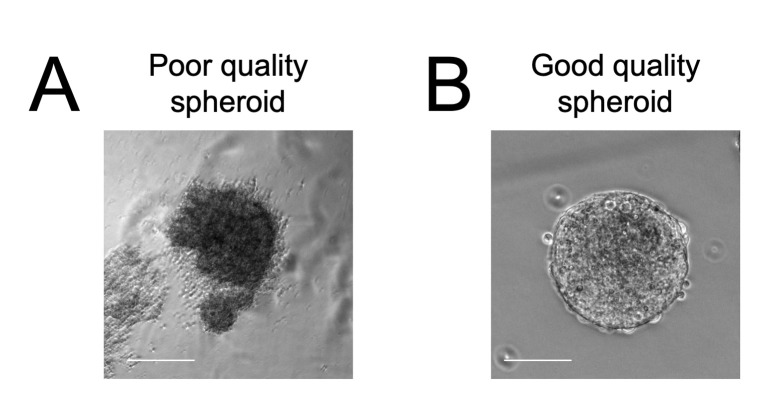
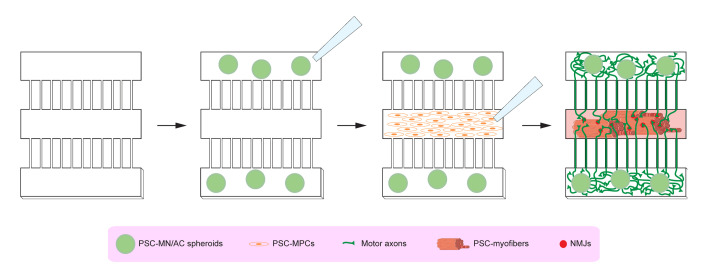
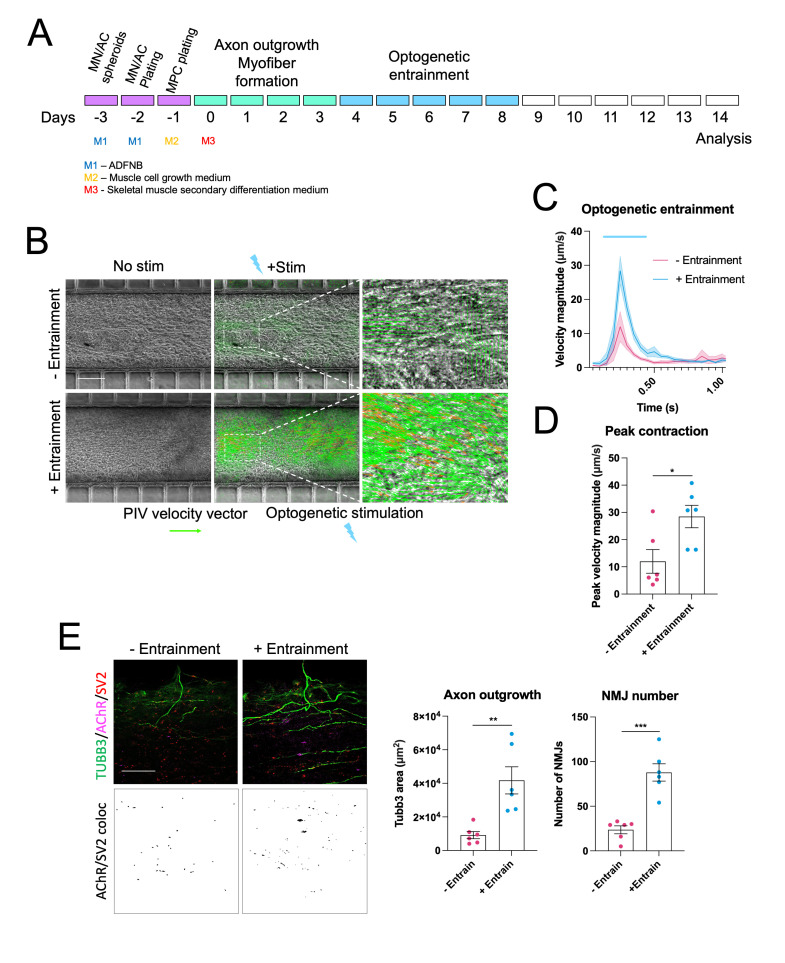
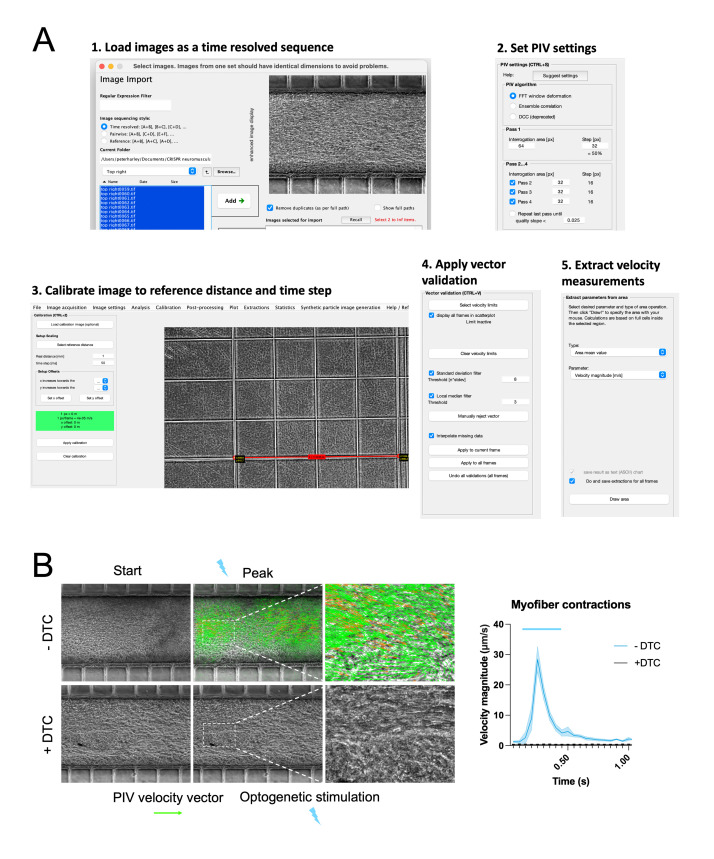
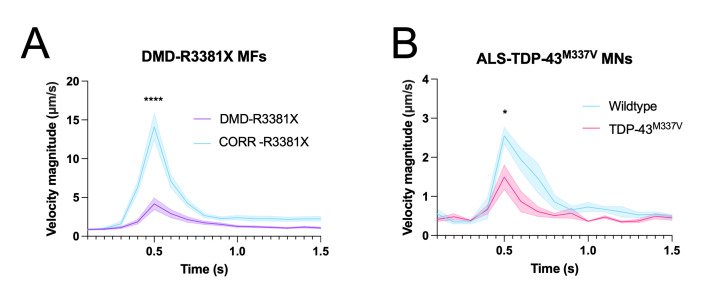
Human neuromuscular diseases represent a diverse group of disorders with unmet clinical need, ranging from muscular dystrophies, such as Duchenne muscular dystrophy (DMD), to neurodegenerative disorders, such as amyotrophic lateral sclerosis (ALS). In many of these conditions, axonal and neuromuscular synapse dysfunction have been implicated as crucial pathological events, highlighting the need for in vitro disease models that accurately recapitulate these aspects of human neuromuscular physiology. The protocol reported here describes the co-culture of neural spheroids composed of human pluripotent stem cell (PSC)-derived motor neurons and astrocytes, and human PSC-derived myofibers in 3D compartmentalised microdevices to generate functional human neuromuscular circuits in vitro. In this microphysiological model, motor axons project from a central nervous system (CNS)-like compartment along microchannels to innervate skeletal myofibers plated in a separate muscle compartment. This mimics the spatial organization of neuromuscular circuits in vivo. Optogenetics, particle image velocimetry (PIV) analysis, and immunocytochemistry are used to control, record, and quantify functional neuromuscular transmission, axonal outgrowth, and neuromuscular synapse number and morphology. This approach has been applied to study disease-specific phenotypes for DMD and ALS by incorporating patient-derived and CRISPR-corrected human PSC-derived motor neurons and skeletal myogenic progenitors into the model, as well as testing candidate drugs for rescuing pathological phenotypes. The main advantages of this approach are: i) its simple design; ii) the in vivo-like anatomical separation between CNS and peripheral muscle; and iii) the amenability of the approach to high power imaging. This opens up the possibility for carrying out live axonal transport and synaptic imaging assays in future studies, in addition to the applications reported in this study. Graphical abstract Channelrhodopsin-2 (CHR2+), pluripotent stem cell (PSC), motor neurons (MNs), myofibers (MFs), neuromuscular junction (NMJ).
人类神经肌肉疾病是一组有着未满足临床需求的多样疾病,从诸如杜氏肌营养不良症(DMD)等肌肉营养不良症,到诸如肌萎缩侧索硬化症(ALS)等神经退行性疾病。在许多这类病症中,轴突和神经肌肉突触功能障碍被认为是关键的病理事件,这凸显了对能准确重现人类神经肌肉生理学这些方面的体外疾病模型的需求。本文报道的方案描述了由人多能干细胞(PSC)衍生的运动神经元和星形胶质细胞组成的神经球与3D分隔微装置中的人PSC衍生的肌纤维的共培养,以在体外生成功能性人类神经肌肉回路。在这个微生理模型中,运动轴突从类似中枢神经系统(CNS)的隔室沿着微通道伸出,以支配置于单独肌肉隔室中的骨骼肌纤维。这模拟了体内神经肌肉回路的空间组织。光遗传学、粒子图像测速(PIV)分析和免疫细胞化学被用于控制、记录和量化功能性神经肌肉传递、轴突生长以及神经肌肉突触数量和形态。通过将患者来源的和经CRISPR校正的人PSC衍生的运动神经元及骨骼肌生成祖细胞纳入模型,以及测试用于挽救病理表型的候选药物,这种方法已被应用于研究DMD和ALS的疾病特异性表型。这种方法的主要优点是:i)其设计简单;ii)CNS与外周肌肉之间类似体内的解剖学分离;iii)该方法适用于高分辨率成像。除了本研究报道的应用外,这为未来研究中进行实时轴突运输和突触成像分析开辟了可能性。图形摘要 通道视紫红质-2(CHR2+)、多能干细胞(PSC)、运动神经元(MNs)、肌纤维(MFs)、神经肌肉接头(NMJ)