Gao Ruimin, Duceppe Marc-Olivier, Chattaway Marie Anne, Goodridge Lawrence, Ogunremi Dele
Ottawa Laboratory Fallowfield, Canadian Food Inspection Agency, Ottawa, ON, Canada.
Department of Food Science and Agricultural Chemistry, McGill University, Ste Anne de Bellevue, QC, Canada.
Front Microbiol. 2023 Mar 3;14:1086198. doi: 10.3389/fmicb.2023.1086198. eCollection 2023.
Outbreak investigation of foodborne salmonellosis is hindered when the food source is contaminated by multiple strains of , creating difficulties matching an incriminated organism recovered from patients with the specific strain in the suspect food. An outbreak of the rare Adjame was caused by multiple strains of the organism as revealed by single-nucleotide polymorphism (SNP) variation. The use of highly discriminatory prophage analysis to characterize strains of should enable a more precise strain characterization and aid the investigation of foodborne salmonellosis.
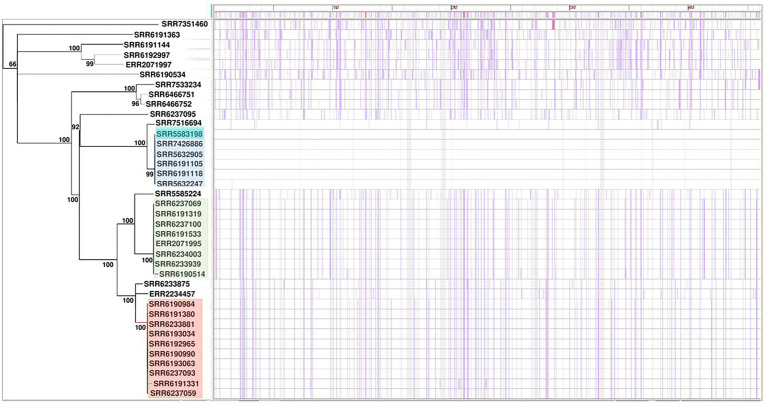
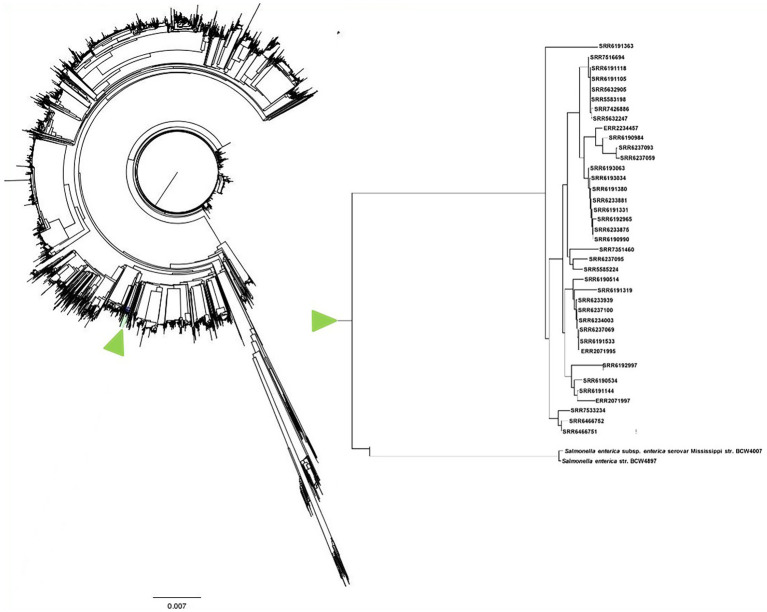
We have carried out genomic analysis of Adjame strains recovered during the course of a recent outbreak and compared them with other strains of the organism ( = 38 strains), using SNPs to evaluate strain differences present in the core genome, and prophage sequence typing (PST) to evaluate the accessory genome. Phylogenetic analyses were performed using both total prophage content and conserved prophages.
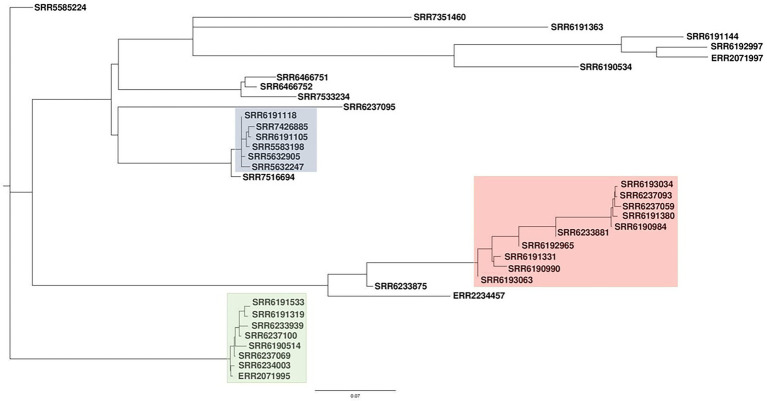
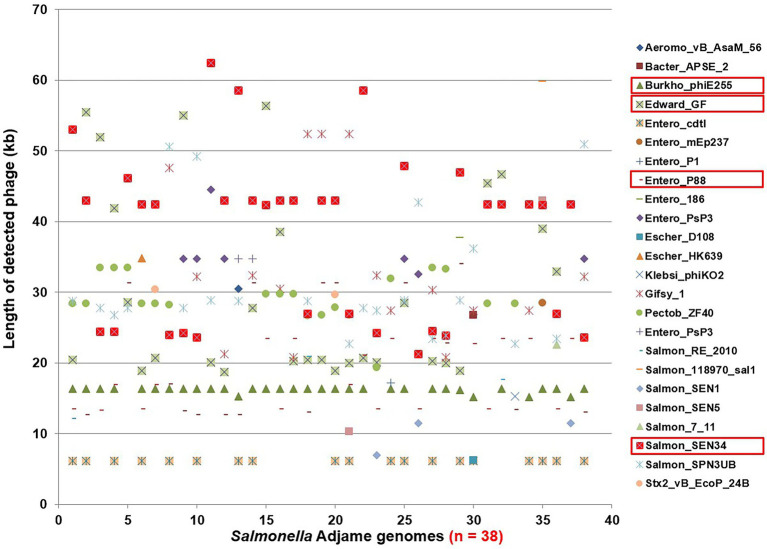
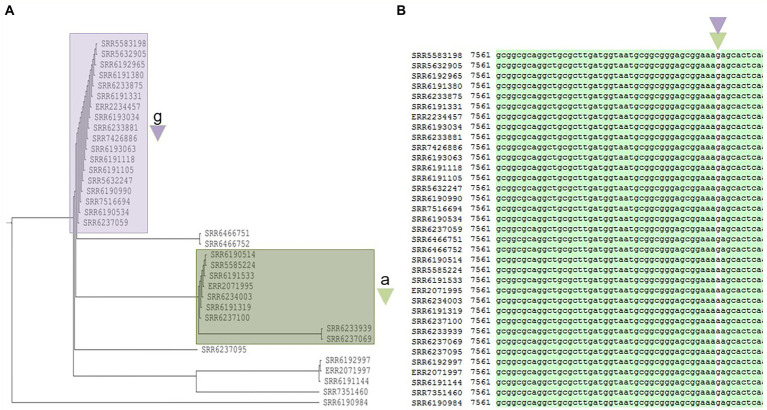
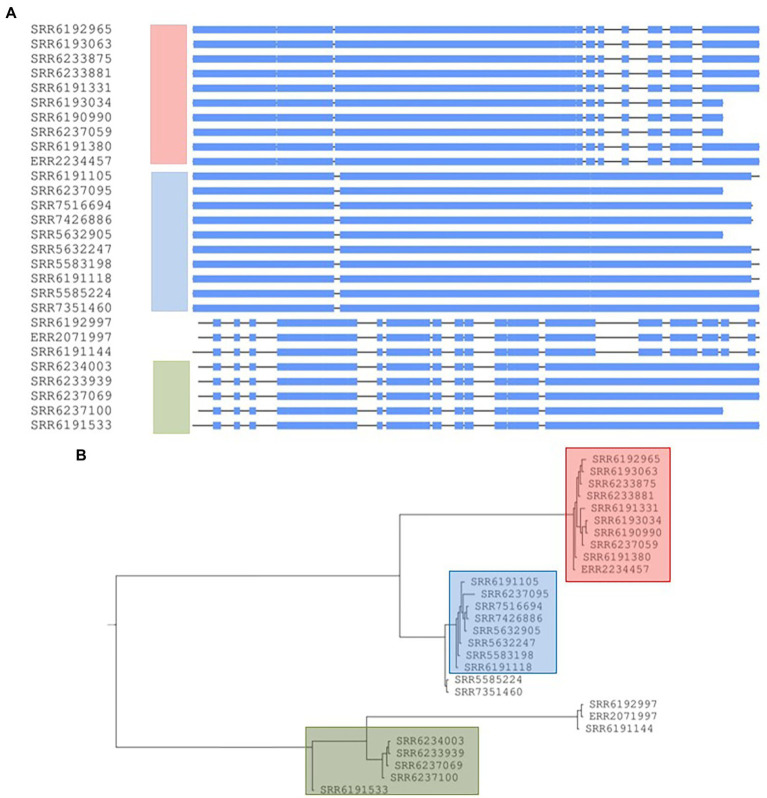
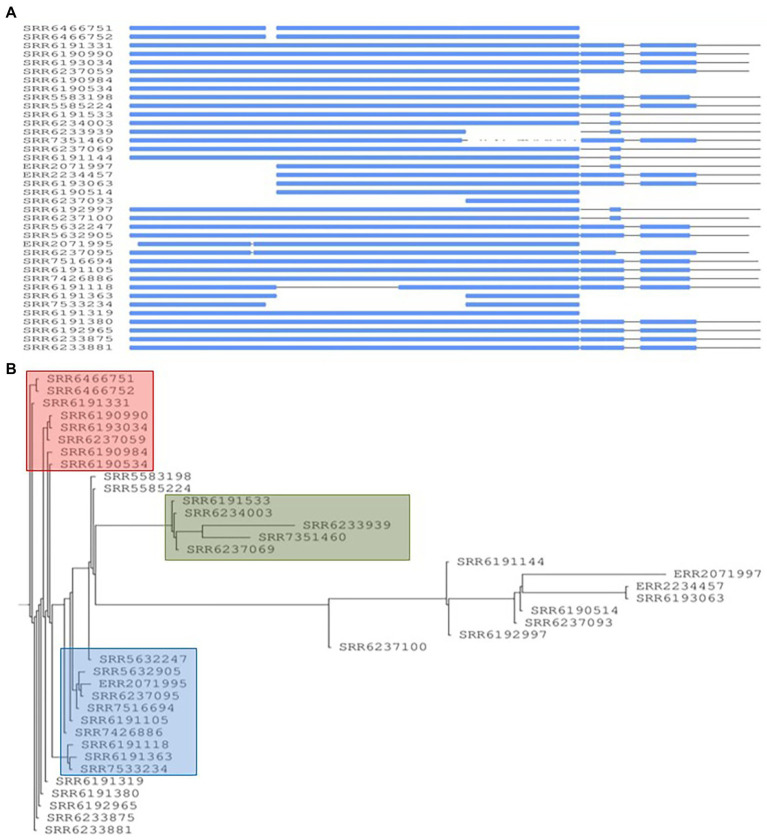
The PST analysis of the Adjame isolates showed a high degree of strain heterogeneity. We observed small clusters made up of 2-6 isolates ( = 27) and singletons ( = 11) in stark contrast with the three clusters observed by SNP analysis. In total, we detected 24 prophages of which only four were highly prevalent, namely: Entero_p88 (36/38 strains), Salmon_SEN34 (35/38 strains), Burkho_phiE255 (33/38 strains) and Edward_GF (28/38 strains). Despite the marked strain diversity seen with prophage analysis, the distribution of the four most common prophages matched the clustering observed using core genome.
Mutations in the core and accessory genomes of Adjame have shed light on the evolutionary relationships among the Adjame strains and demonstrated a convergence of the variations observed in both fractions of the genome. We conclude that core and accessory genomes analyses should be adopted in foodborne bacteria outbreak investigations to provide a more accurate strain description and facilitate reliable matching of isolates from patients and incriminated food sources. The outcomes should translate to a better understanding of the microbial population structure and an 46 improved source attribution in foodborne illnesses.
当食物来源被多种菌株污染时,食源性沙门氏菌病的暴发调查会受到阻碍,这使得从患者身上分离出的致病生物体难以与可疑食物中的特定菌株相匹配。如单核苷酸多态性(SNP)变异所揭示的,罕见的阿贾梅沙门氏菌的一次暴发是由该生物体的多种菌株引起的。使用高度有鉴别力的前噬菌体分析来鉴定沙门氏菌菌株应能实现更精确的菌株鉴定,并有助于食源性沙门氏菌病的调查。
我们对近期一次暴发过程中分离出的阿贾梅沙门氏菌菌株进行了基因组分析,并将它们与该生物体的其他菌株(n = 38株)进行比较,使用SNP评估核心基因组中存在的菌株差异,并使用前噬菌体序列分型(PST)评估辅助基因组。使用总前噬菌体含量和保守前噬菌体进行系统发育分析。
对阿贾梅分离株的PST分析显示出高度的菌株异质性。我们观察到由2 - 6个分离株组成的小簇(n = 27)和单株(n = 11),这与SNP分析观察到的三个簇形成鲜明对比。我们总共检测到24种前噬菌体,其中只有四种高度普遍,即:Entero_p88(36/38株)、Salmon_SEN34(35/38株)、Burkho_phiE255(33/38株)和Edward_GF(28/38株)。尽管在前噬菌体分析中观察到明显的菌株多样性,但四种最常见前噬菌体的分布与使用核心基因组观察到的聚类相匹配。
阿贾梅沙门氏菌核心基因组和辅助基因组中的突变揭示了阿贾梅菌株之间的进化关系,并证明了在基因组的两个部分中观察到的变异趋同。我们得出结论,在食源性细菌暴发调查中应采用核心基因组和辅助基因组分析,以提供更准确的菌株描述,并促进患者分离株与可疑食物来源的可靠匹配。结果应有助于更好地理解微生物种群结构,并改善食源性疾病的来源归因。